Research Article - (2013) Volume 4, Issue 11
The present study investigated the combined effect of fenofibrate (PPAR-α agonist) and saxagliptin (DPP-4 inhibitor) in diabetes-induced experimental nephropathy. Rats were administered streptozotocin (STZ) (55 mg/ kg i.p., once) to induce experimental diabetes mellitus. The development of diabetic nephropathy was assessed biochemically and histologically. In addition, the diabetes-induced lipid profile and renal oxidative stress were assessed. The single administration of STZ produced diabetes, which induced renal oxidative stress, altered the lipid profile and subsequently produced nephropathy in 8 weeks by increasing serum creatinine, blood urea nitrogen, proteinuria, and glomerular damage. Treatment with fenofibrate (30 mg/kg/day p.o.) normalized the altered lipid profile in diabetic rats, whereas the saxagliptin (3 mg/kg p.o.) treatment has no effect on lipid alteration in diabetic rats. Treatment with saxagliptin partially reduces the elevated glucose levels in diabetic rats, whereas fenofibrate treatment has no effect on it. The combination of fenofibrate and saxagliptin was more effective in attenuating the diabetes-induced nephropathy and renal oxidative stress as compared to treatment with either drug alone or lisinopril (1 mg/kg/day p.o.) (a standard agent). It may be concluded that diabetes-induced oxidative stress and lipid alterations may be responsible for the induction of nephropathy in diabetic rats. The concurrent administration of fenofibrate and saxagliptin may have prevented the development of diabetes-induced nephropathy by reducing the lipid alteration, decreasing the renal oxidative stress and providing the direct nephroprotective action.
Keywords: Diabetes; DPP-4 inhibitor; Oxidative stress; Nephropathy; PPAR- α agonist
Diabetes mellitus, a metabolic syndrome is known to trigger various complications including retinopathy, neuropathy, nephropathy etc. Diabetic nephropathy is an important microvascular complication of diabetes and is widely recognized as the most common cause of the end-stage renal failure. All the kidney cellular elements, i.e., glomerular endothelia, mesangial cells, podocytes, and tubular epithelia, are targets of hyperglycaemic injury. Glomerulosclerosis, thickening of the glomerular basement membrane, glomerular hypertrophy, mesangial cell expansion, podocyte loss, renal-cell hypertrophy and tubulointerstitial fibrosis are major pathological changes during the course of diabetic nephropathy, which ultimately results in functional consequences, including progressive albuminuria, reduction in glomerular filtration rate, elevation of arterial blood pressure and fluid retention [1-5]. Numerous studies have suggested that hyperglycaemia induced hemodynamic pathways play a detrimental role in the induction and progression of diabetic nephropathy and are recognized to be major mediators of kidney disease [6-8], but diabetes-induced lipid profile alteration has been noted to be an independent and major factor in the progression of diabetes-induced nephropathy. Altered lipid profile during the diabetic state has been noted to be associated with the increased expression of transforming growth factor- β1 (TGF-β1), connective tissue growth factor (CTGF), fibronectin, collagen IV, mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB) which ultimately accounts for glomerulosclerosis and tubulointerstitial fibrosis [9]. Recently, it has demonstrated that renal lipid accumulation is associated with the increased renal biglycan content which contributes to the development of diabetic nephropathy [10]. The chronic diabetes mellitus may mediate renal injury by increasing the renal expression of sterol regulatory element-binding protein (SREBP-1), which is responsible for increasing the synthesis of triglycerides and cholesterol in the kidney, that are associated with upregulation of TGF-β and extracellular matrix proteins, resulting in glomerulosclerosis and tubulointerstitial fibrosis [11]. Increased expression of TGF-β has been noted to be implicated in pathogenesis of diabetic nephropathy by increasing the PAI-1 synthesis, stimulation of fibronectin synthesis in mesangial cells, decreasing the activity of plasminogen activator (PA) and MMP-2, stimulating the expression of CTGF, which promotes the renal deposition of extracellular matrix components like collagen I & IV, stimulation of VEGF expression and by enhancing the expression of interleukin-18 (IL-18) in human renal proximal tubular epithelial cells. Interestingly, elevated levels of LDL have been shown to increase the expression of TGF-β, CTGF and collagen-I in human mesangial cells [12,13]. Thus, it is mandatory to develop novel therapeutic interventions to prevent dyslipidemiaassociated induction and progression of nephropathy in patients with diabetes mellitus. Peroxisome proliferator activated receptors-alpha (PPAR-α), a ligand-activated transcription factor of nuclear hormone receptor superfamily, plays an important role in the oxidation of fatty acids and activation of PPAR-α promotes lipid metabolism [14]. Recently, it has been shown that treatment with fenofibrate partially prevented the development of nephropathy by reducing albuminuria, serum creatinine and blood urea nitrogen and preventing the glomerular damage in diabetic rats [15].
Glucagon-like peptide 1 (GLP-1) is a gut-derived incretin hormone. GLP-1 stimulates insulin secretion, suppresses glucagon secretion, delays gastric emptying, and inhibits small bowel motility, all actions contributing to the anti-diabetogenic effect. But, the GLP-1 is rapidly inactivated by the dipeptidyl peptidase-4 (DPP-4) enzyme, which results in a short circulating half-life of the active form of GLP-1. Thereby, therapeutic approaches for enhancing incretin action include degradation-resistant GLP-1 receptor agonists (incretin mimetics), and inhibitors of DPP-4 activity (incretin enhancers). The both approaches have been found to be promising in lowering the glucose level in the experimental as well as clinical studies. It is interesting to note that GLP-1 expression has also been found in the kidney of rats and the rGLP-1 found to inhibit the Na+ reabsorption in the proximal tubule and increases glomerular filtration rate in kidneys [16,17]. In addition, GLP-1 has been noted to improve endothelial dysfunction in type 2 diabetic patients with coronary heart disease [18]. Moreover, treatment with exendin-4 (GLP-1 receptor agonists) showed a significant reduction in glomerular hypertrophy, mesangial matrix expansion, TGF-β1 expression, and type IV collagen accumulation and associated glomerular lipid accumulation in db/db mice [19]. Therefore, simultaneous activation of PPAR-α and GLP-1 by inhibitors of DPP- 4 activity (incretin enhancers) could be a potential therapeutic option to prevent diabetes-associated hyperlipidemia and hyperglycemia– provoked induction and progression of diabetic nephropathy. Therefore the present study has been designed to investigate the effect of combination of PPAR-α agonist and DPP-4 inhibitor in diabetesinduced nephropathy in rats.
The experimental protocol used in the present study was approved by the Institutional Animal Ethical Committee. Age matched young wistar rats weighing about 200-240 g were employed in the present study. Rats were fed on standard chow diet and water ad libitum. They were acclimatized in institutional animal house and were exposed to normal cycles of day and night.
Assessment of diabetes and lipid profile
The experimental diabetes mellitus was induced in rats by single injection of streptozotocin (STZ) (55 mg/kg i.p.,) dissolved in freshly prepared ice cold citrate buffer of pH 4.5. The blood sugar level was monitored once daily for first week after administration of STZ. Then, at the end of the experimental protocol (8 weeks after administration of STZ), the blood samples were collected and serum was separated. The serum samples were frozen until analysing the biochemical parameters. The serum glucose concentration was estimated by glucose oxidase peroxidase (GOD-POD) method [20] using the commercially available kit (Transasia Bio-Medical Ltd., Solan, India). The serum total cholesterol was estimated by cholesterol oxidase peroxidase (CHOD/PAP) method [21] using the commercially available kit (Transasia Bio-Medical Ltd., Solan, India). The serum triglyceride was estimated by glycerol phosphate oxidase (GPO/PAP) method [22] using the commercially available kit (Transasia Bio-Medical Ltd., Solan, India). The serum high density lipoprotein (HDL) was estimated by polyethylene glycol (PEG) precipitation method [21] using the commercially available kit (Transasia Bio-Medical Ltd., Solan, India).
Assessment of diabetic nephropathy
The diabetes mellitus-induced nephropathy was assessed biochemically by estimating serum creatinine, blood urea nitrogen and proteinuria.
Estimation of serum creatinine: The serum creatinine concentration was estimated by alkaline picrate method [23] using the commercially available kit (Cresent biosystems, Goa, India). Briefly, 2.0 ml of picric acid reagent was added to 0.2 ml of serum for deproteinization of specimen, which was mixed well and centrifuged at 3000 rpm to obtain a clear supernatant. 100 μl of buffer reagent was added to 1.1 ml of supernatant, 0.1 ml of standard creatinine and 0.1 ml of distilled water to prepare test, standard and blank, respectively. 1.0 ml of picric acid reagent was added to blank and standard. The test tubes were mixed well and kept at room temperature for 20 minutes. The alkaline picrate reacts with creatinine to form the orange coloured complex, which was read at 520 nm spectophotometrically. The serum creatinine concentration was calculated using the following formula:
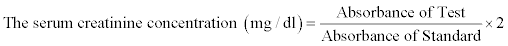
Estimation of blood urea nitrogen: The blood urea was estimated by Berthelot method [24] using the commercially available kit (Kamineni Life Sciences Pvt. Ltd. Hyderabad, India). 1000 μl of working reagent-I containing urease reagent, and a mixture of salicylate, hypochlorite and nitroprusside was added to 10 μl of serum, 10 μl of standard urea (40 mg/dl) and 10 μl of purified water to prepare test, standard and blank, respectively. All the test tubes were mixed well and incubated at 37°C for 5 min. Then 1000 μl of reagent-II containing alkaline buffer, was added to all the test tubes, which were incubated at 37°C for 5 min. Urease catalyses the conversion of urea to ammonia and carbon dioxide. The ammonia thus released reacts with a mixture of salicylate, hypochlorite and nitroprusside to yield indophenol, a blue-green coloured compound. The intensity of the colour produced is directly proportional to the concentration of urea in the sample and is measured spectrophotometrically at 578 nm. The blood urea was calculated using the following formula:
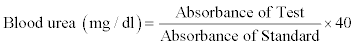
Blood urea nitrogen (mg/dl)=Serum urea × 0.467
Estimation of protein in urine: The proteinuria was assessed by pyrogallol red method [25] using the commercially available kit (Transasia Bio-Medical Ltd., Solan, India). 1000 μl of reagent (pyragallol dye) was added to 10 μl of urine sample, 10 μl of standard protein and 10 μl of purified water to prepare test, standard and blank, respectively. All the test tubes were mixed and incubated at 37°C for 10 min. The absorbances of test and standard samples were noted against blank at 600 nm spectophotometrically. When the pyrogallol red-molybedate complex binds to basic amino groups of protein molecules, there is a shift in reagent absorbance. The absorbance is directly proportional to protein concentration present in the sample. The urinary protein was calculated using the following formula:
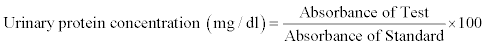
Total microprotein excreted (mg/24 hours)=Urinary protein concentration (mg/dl) ×10×total volume of urine (liters) excreted for 24 hours
Histopathological study: The early diabetic changes in glomeruli were assessed histologically as previously described [26,27]. The kidney was excised and immediately immersed in 10% formalin. The kidney was dehydrated in graded concentrations of alcohol, immersed in xylene and then embedded in paraffin. From the paraffin blocks, sections of 3 μm in thickness were made and stained with hematoxylin and eosin to assess the pathological changes occurs in glomeruli using light microscopy (40X).
Assessment of renal oxidative stress
The development of oxidative stress in the kidney was assessed by estimating renal thiobarbituric acid reactive substances (TBARS) and reduced form glutathione (GSH).
Preparation of renal homogenate: The kidney was dissected and washed with ice cold isotonic saline and weighed. The kidney was then minced, and a homogenate (10% w/v) was prepared in chilled 1.15% KCl. The homogenate was used for estimating TBARS, GSH and total protein.
Estimation of TBARS: The renal TBARS, an index of lipid peroxidation, was estimated according to the method described earlier [28]. The reaction mixture was prepared by mixing 0.2 ml of tissue homogenate, 0.2 ml of 8.1% sodium dodecyl sulphate (SDS), 1.5 ml of 20% acetic acid solution adjusted to pH 3.5 with NaOH, and 1.5 ml of 0.8% aqueous solution of thiobarbituric acid (TBA). The reaction mixture was made up to 4.0 ml with distilled water, and then heated in water bath at 95°C for 60 min. After cooling in tap water, 1.0 ml of distilled water and 5.0 ml of the mixture of n-butanol and pyridine (15:1 v/v) were added to reaction mixture and shaken vigorously. After centrifugation at 4000 rpm for 10 min, the organic layer was taken and its absorbance at 532 nm was measured. The standard curve using 1, 1, 3, 3-tertramethoxyopropane was plotted to calculate the concentration of TBARS and the results were expressed as nmol/mg of protein.
Estimation of reduced glutathione: The GSH level in the kidney was estimated using the method described by Ellman [29]. Briefly, the renal homogenate was mixed with 10% w/v trichloroacetic acid in ratio of 1:1 and centrifuged at 4°C for 10 min at 5000 rpm. The supernatant obtained (0.5 ml) was mixed with 2 ml of 0.3 M disodium hydrogen phosphate buffer (pH 8.4) and 0.4 ml of distilled water. Then 0.25 ml of 0.001 M freshly prepared DTNB (5, 5’-dithiobis (2-nitrobenzoic acid) dissolved in 1% w/v sodium citrate) was added. The reaction mixture was incubated for 10 min and absorbance of yellow coloured complex was noted spectrophotometrically at 412 nm. A standard curve was plotted using reduced form of glutathione and the results were expressed as nmol/mg of protein.
Estimation of total protein: The renal total protein content was estimated by Lowry’s method [30] using bovine serum albumin (BSA) as a standard. Briefly, 0.3 ml of tissue homogenate was diluted to 1 ml. The 100 μl of diluted supernatant was made up to 1 ml using distilled water. To this, 5 ml of Lowry’s reagent was added. The contents were mixed thoroughly and the mixture was allowed to stand for 15 min at room temperature. Then 0.5 ml of Folin-ciocalteu reagent was added and the contents were vortexed vigorously and incubated at room temperature for 30 min. The standard curve was plotted using BSA. The protein content was determined spectrophotometrically at 750 nm.
Experimental protocol
Eight groups were employed in the present study and each group comprised 6 rats. The fenofibrate and saxagliptin were suspended in 0.5% w/v of carboxy methyl cellulose (CMC). Group I (Normal Control), rats were maintained on standard food and water and no treatment was given. Group II (Diabetic Control), rats were administered STZ (55 mg/ kg, i.p., once) dissolved in citrate buffer (pH 4.5). Group III (Fenofibrate per se), the normal rats were administered fenofibrate (30 mg/kg p.o.) suspended in 0.5% w/v of CMC for 7 weeks. Group IV (Saxagliptin per se), the normal rats were administered saxagliptin (3 mg/kg p.o.) suspended in 0.5% w/v of CMC for 7 weeks. Group V (Fenofibrate Treated), the diabetic rats, after 1 week of STZ administration, were treated with low dose of fenofibrate (30 mg/kg p.o.) for 7 weeks. Group VI (Saxagliptin Treated), the diabetic rats, after 1 week of STZ administration, were treated with saxagliptin (3 mg/ kg p.o.) for 7 weeks. Group VII (Fenofibrate + Saxagliptin Treated), the diabetic rats, after 1 week of STZ administration, were treated with the combination of low dose of fenofibrate (30 mg/kg, p.o.) and saxagliptin (3 mg/kg p.o.) for 7 weeks. Group VIII (Lisinopril Treated), the diabetic rats after 1 week of STZ administration, were treated with lisinopril (1 mg/kg p.o.) for 7 weeks.
Statistical analysis
All values were expressed as mean ± SD. The data obtained from various groups were statistically analyzed using one way ANOVA, followed by Tukey’s multiple comparison tests. The p value of less than 0.05 was considered to be statistically significant and the p values were of two tailed.
Drugs and chemicals
Streptozotocin was obtained from Sigma-Aldrich Ltd., St. Louis, USA. 1, 1, 3, 3-tetra methoxypropane and carboxymethyl cellulose were purchased from R. K. Enterprises, Meerut, India. Fenofibrate and saxagliptin were obtained from Ranbaxy Laboratory Ltd., Gurgaon, India. Saxagliptin was obtained from Bristol-Myers Squibb, Mumbai, India. Lisinopril was obtained from Dr. Reddy’s Laboratory Ltd., Hyderabad, India. All other chemicals used in the present study were of analytical grade.
Administration of fenofibrate at lower dose (30 mg/kg p.o., 7 weeks) or saxagliptin (3 mg/kg p.o., 7 weeks) did not produce any significant per se effect on various parameters assessed in normal rats. Administration of streptozotocin (STZ) (55 mg/kg, i.p., once) produced hyperglycemia after 72 h (serum glucose >180mg/dL). After 7 days of STZ administration, the rats showed blood glucose level of greater than 270 mg/dL were selected and were named as diabetic rats. Fenofibrate (30 mg/kg p.o., 7 weeks), saxagliptin (3 mg/kg p.o., 7 weeks) and lisinopril (1 mg/kg, p.o., 7 weeks) were administered to diabetic rats after 7 days of single injection of STZ and their treatments were continued for 7 weeks. All the parameters were assessed at the end of 7 weeks in normal and diabetic rats with or without drug treatments. Less than 12% of mortality rate was observed in diabetic rats with or without drug treatments.
Effect of pharmacological interventions on serum glucose
The marked increase in serum concentration of glucose was noted in diabetic rats as compared to normal rats. Treatment with fenofibrate (30 mg/kg p.o., 7 weeks) did not affect the serum glucose concentration in diabetic rats. However, treatment with saxagliptin (3 mg/kg p.o., 7 weeks) incompletely reduced the elevated glucose level in diabetic rats. In addition, the effect of saxagliptin in incompletely reduction of elevated serum glucose level in diabetic rats was not altered by its combination with fenofibrate (30 mg/kg p.o., 7 weeks). The lisinopril (1 mg/kg p.o., 7 weeks) treatment slightly lowered the glucose level, but the results were not statistically significant (Table 1).
| Assessments | Normal Control | Diabetic Control | Feno per se | Saxa per se | Feno Treated Diabetic Group | Saxa Treated Diabetic Group | Feno + Saxa Treated Diabetic Group | Lisino Treated Diabetic Group |
|---|---|---|---|---|---|---|---|---|
| Glucose (mg/dl) | 101.93 ± 10.88 | 367.98 ± 35.68a | 101.96 ± 14.98 | 104.10 ± 11.28 | 354.23 ± 40.72 | 299.98 ± 34.17b | 294.98 ± 32.56b | 336.73 ± 36.72 |
| Total Cholesterol (mg/dl) | 58.08 ± 7.41 | 86.06 ± 11.21a | 58.67 ± 6.42 | 57.15 ± 6.72 | 65.16 ± 7.62b | 83.58 ± 9.56 | 64.31 ± 8.36b | 81.08 ± 10.50 |
| Triglycerides (mg/dl) | 95.75 ± 10.10 | 189.29 ± 19.20a | 95.32 ± 12.82 | 95..81 ± 11.74 | 122.15 ± 17.53b | 178.31 ± 19.39 | 115.38 ± 16.83b | 158.75 ± 16.04b |
| High Density Lipoproteins (mg/dl) | 35.81 ± 3.86 | 26.18 ± 3.27a | 36.83 ± 4.28 | 36.23 ± 3.34 | 35.05 ± 4.35b | 27.99 ± 3.67 | 37.77 ± 4.32b | 30.39 ± 2.85 |
Table 1: Effect of fenofibrate (feno), saxagliptin (saxa) and lisinopril on serum glucose and lipid profile. All values are represented as mean ± SD. a=p< 0.05 versus normal control; b=p< 0.05 versus diabetic control.
Effect of pharmacological interventions on serum lipid profile
The increase in serum concentrations of total cholesterol and triglycerides and consequent decrease in HDL levels were noted in diabetic rats as compared to normal rats. Treatment with fenofibrate (30 mg/kg p.o., 7 weeks) significantly attenuated diabetes-induced alteration in lipid levels. However, treatment with saxagliptin (3 mg/ kg p.o., 7 weeks) did not affect the lipid alterations in diabetic rats. In addition, the effect of fenofibrate in attenuating the lipid alterations in diabetic rats was not enhanced by its combination with saxagliptin (3 mg/kg p.o., 7 weeks). On the other hand, treatment with lisinopril (1 mg/kg, p.o., 7 weeks) partially prevented the diabetes-induced increases in total cholesterol and triglycerides and consequent decrease in HDL levels; but results were significant only in case of triglycerides (Table 1).
Effect of pharmacological interventions on serum creatinine and blood urea nitrogen
The serum creatinine and blood urea nitrogen levels were noted to be markedly increased in diabetic rats as compared to normal rats. Treatment with either fenofibrate (30 mg/kg p.o., 7 weeks) or saxagliptin (3 mg/kg p.o., 7 weeks) partially reduced the diabetes-induced increases in serum creatinine and blood urea nitrogen. Moreover, the concurrent administration of fenofibrate (30 mg/kg p.o., 7 weeks) and saxagliptin (3 mg/kg p.o., 7 weeks) markedly reduced the elevated levels of serum creatinine and blood urea nitrogen as compared to treatments with either drug alone or lisinopril (1 mg/kg, p.o., 7 weeks) in diabetic rats (Figures 1 and 2).

Figure 1: Effect of fenofibrate (feno) and saxagliptin (saxa) on serum creatinine. All values are represented as mean ± SD. a=p< 0.05 versus normal control; b=p< 0.05 versus diabetic control; c=p<0.05 versus fenofibrate treated diabetic group; d=p< 0.05 versus saxagliptin treated diabetic group; e=P<0.05 versus lisinopril treated diabetic group.

Figure 2: Effect of fenofibrate (feno) and saxagliptin (saxa) on Blood urea nitrogen. All values are represented as mean ± SD. a=p<0.05 versus normal control; b=p< 0.05 versus diabetic control; c=p<0.05 versus fenofibrate treated diabetic group; d= p<0.05 versus saxagliptin treated diabetic group; e=P<0.05 versus lisinopril treated diabetic group.
Effect of pharmacological interventions on proteinuria
A marked increase in the occurrence of proteinuria was noted in diabetic rats as compared to normal rats. Treatment with either fenofibrate (30 mg/kg p.o., 7 weeks) or saxagliptin (3 mg/kg p.o., 7 weeks) partially reduced the incidence of proteinuria in diabetic rats. Moreover, the concurrent administration of fenofibrate (30 mg/kg p.o., 7 weeks) and saxagliptin (3 mg/kg p.o., 7 weeks) markedly reduced the incidence of proteinuria as compared to treatments with either drug alone or lisinopril (1 mg/kg, p.o., 7 weeks) in diabetic rats (Figure 3).

Figure 3: Effect of fenofibrate (feno) and saxagliptin (saxa) on urinary protein. All values are represented as mean ± SD. a=p<0.05 versus normal control; b=p<0.05 versus diabetic control; c=p<0.05 versus fenofibrate treated diabetic group; d = p< 0.05 versus saxagliptin treated diabetic group; e=P<0.05 versus lisinopril treated diabetic group.
Effect of pharmacological interventions on renal oxidative stress
Diabetic rats after 7 weeks (8 weeks after STZ administration) showed marked increase in renal TBARS as compared to normal rats. In addition, the renal concentration of GSH was noted to be decreased in diabetic rats as compared to normal rats. Treatment with either fenofibrate (30 mg/kg p.o., 7 weeks) or saxagliptin (3 mg/ kg p.o., 7 weeks) partially prevented the diabetes-induced increase in renal TBARS and decrease in renal GSH. Moreover, the concurrent administration of fenofibrate (30 mg/kg p.o., 7 weeks) and saxagliptin (3 mg/kg p.o., 7 weeks) markedly attenuated the diabetes-induced increase in renal TBARS and decrease in renal GSH as compared to treatments with either drug alone orc (1 mg/kg, p.o., 7 weeks) in diabetic rats (Figures 4 and 5).

Figure 4: Effect of fenofibrate (feno) and saxagliptin (saxa) on renal TBAR. All values are represented as mean ± SD. a=p<0.05 versus normal control; b=p<0.05 versus diabetic control; c=p<0.05 versus fenofibrate treated diabetic group; d = p< 0.05 versus saxagliptin treated diabetic group; e=P<0.05 versus lisinopril treated diabetic group.

Figure 5: Effect of fenofibrate (feno) and saxagliptin (saxa) on Renal GSH. All values are represented as mean ± SD. a=p<0.05 versus normal control; b=p<0.05 versus diabetic control; c=p<0.05 versus fenofibrate treated diabetic group; d=p<0.05 versus saxagliptin treated diabetic group; e=P<0.05 versus lisinopril treated diabetic group.
Effect of pharmacological interventions on renal histological study
The diabetic rats were noted to develop pathological changes in the glomeruli such as glomerular capillary size reduction and extracellular mesangial expansion after 7 weeks as compared to normal rats. Treatment with either fenofibrate (30 mg/kg p.o., 7 weeks) or saxagliptin (3 mg/kg p.o., 7 weeks) partially prevented the diabetes-induced pathological changes in the glomeruli by improving the glomerular capillary size and reducing the mesangial expansion in diabetic rats. In addition, the concurrent administration of fenofibrate (30 mg/kg p.o., 7 weeks) and saxagliptin (3 mg/kg p.o., 7 weeks) markedly protected the diabetic kidney from pathological changes in the glomeruli as compared to treatments with either drug alone or lisinopril (1 mg/kg, p.o., 7 weeks) in diabetic rats (Figure 6).

Figure 6: Effect of fenofibrate (feno) and saxagliptin (saxa) on pathological changes in glomeruli. The section of 3 μM in thickness were made and stained with hematoxylin and eosin to assess the pathological changes of glomeruli using the light microscopy (40 X). The diabetic rats were noted to develop pathological changes in the glomeruli such as glomerular capillary size reduction and extracellular mesangial expansion as compared to normal rats. The concurrent administration of fenofibrate and saxagliptin markedly reduced the pathological changes in glomeruli by improving the glomerular capillary size and reducing the mesangial expansion as compared to treatment with either drug alone or lisinopril.
Numerous efforts have been made to identify the major culprit involved in the pathogenesis of this disease and these findings argue that numerous pathways such as hemodynamic pathway involving the renin-angiotensin-aldosterone and urotensin systems, profibrotic and inflammatory cytokines including TGF-β and tumour necrosis factor-α, various kinases such as protein kinase C (PKC) and Janus kinase pathway and most importantly oxidative stress mediator like NADPH oxidase are activated during the course of this disease [31-36]. But, the clinical strategies based on these pathways for the management of diabetic nephropathy remains unsatisfactory, as the numbers of diabetic patients with nephropathy are increasing year-by-year. The present study has been aimed to explore the possible therapeutic strategy to prevent diabetic nephropathy. Our study showed the renoprotective effects of combination of fenofibrate and saxagliptin in diabetes-induced experimental nephropathy. Creatinine, a non-protein waste product is freely filtered by the kidney and as the serum creatinine level depends on the glomerular filtration rate (GFR), thereby is considered to be an index of renal dysfunction [37-39]. Another waste product of protein metabolism namely urea is cleared from the bloodstream by the kidney and it has been considered as an important bio-marker for dysfunction of the kidney [37,40]. Increased cortical interstitial fibrous tissue is correlated with increased mesangial enlargement. Since mesangial expansion was related to albuminuria, GFR, and hypertension, it follows that the index of interstitial fibrosis predicted these clinical manifestations of diabetic nephropathy [41,42]. Altogether, elevated levels of serum creatinine, blood urea nitrogen, and proteinuria, and pathological changes in glomeruli have been recognized as an index of experimental diabetic nephropathy. In the present study, the serum creatinine, blood urea nitrogen, and proteinuria were noted to be increased in diabetic rats in 7 weeks as compared to normal rats. In addition, the diabetic rats showed marked renal pathological changes after 7 weeks. These results clearly signify the development of diabetesinduced nephropathy. Treatment with either fenofibrate or saxagliptin prevented the diabetes-induced nephropathy to a degree by decreasing serum creatinine, blood urea nitrogen and proteinuria and the same effect was observed on renal pathological changes. On the other hand, concurrent administration of fenofibrate and saxagliptin noticeably prevented the development of diabetes-induced nephropathy as compared to treatment with either drug alone.
Diabetic kidneys specifically expressed several genes normally found in adipocytes, including adipose differentiation-related protein (ADRP, or adipophilin in humans), suggesting a switch of kidney phenotype in favour of lipid accumulation in diabetes [43]. In addition, abnormal high concentration of serum lipids is mainly due to increase in the mobilization of free fatty acids from the peripheral fat deposits, because insulin inhibits the hormone sensitive lipase production and this altered lipid profile during the diabetic state has been noted to be associated with the increased expression of TGF-β1, fibronectin, collagen-IV, MAPKs and NF-κB which ultimately accounts for glomerulosclerosis and tubulointerstitial fibrosis [9]. Therefore, Lipid-mediated injury plays an important role in the pathogenesis of many renal diseases, including diabetic nephropathy. This contention is supported by the results obtained in the present study that marked increases in serum cholesterol and triglycerides and consequent decrease in serum HDL levels were noted in diabetic rats with nephropathy. Decreased expression of PPAR-α has been noted to be associated with the progression of diabetic nephropathy through an increase in circulating lipids, induction of inflammation and extracellular matrix formation in diabetic mice [44]. Fenofibrate, one of the most widely used PPAR-α, along with its well established actions on lipids and multiple mechanisms have been postulated regarding the direct renoprotection by decreasing the renal COX-2 expression and reducing the nitrosative stress in the kidney of diabetic rats with early stages of nephropathy [45]. TGF-β plays an important role in mediating the hypertrophic and fibrotic/sclerotic manifestation diabetic nephropathy. Numerous in-vitro as well as in-vivo investigations have shown that renal TGF-β production is increased during the development of diabetic kidney disease by increasing the expression of CTGF, VEGF, collagen I, collagen IV, and fibronectin that results in disassembly and hypertrophy of mesangial cells [46-49], thereby implicated in the pathogenesis of diabetic nephropathy. In addition, TGF-β1 upregulates the expression of p22phox, p47phox, p67phox, and gp91phox in rat mesangial cells and p22phox mRNA in tubular epithelial cells suggesting that TGF-β1-induced ROS may be NADPH oxidase dependent [50]. It is worth-mentioning that activation of PPAR-α by fenofibrate has been reported to produce renoprotective effect by downregulating the renal expression of TGF-β [51]. These studies explicate the possible mechanisms involved in fenofibrate-mediated renoprotective effects noted in the present study.
Hyperglycemia is an important contributor for the cardiovascular diseases risk. The animal studies revealed that hyperglycemia produces glycation and peroxidation of proteins which cause damage to the vascular walls [52]. Uncontrolled hyperglycemia plays an important role in the induction and progression of nephropathy by upregulating the renal expression of Ang-II, PKC, TGF-β and other intermediary growth factors [53]. Moreover, high glucose-induced ROS generation through an activation of NADPH oxidase play a crucial role in induction and progression of diabetic nephropathy [5]. GLP-1 receptor agonists (incretin mimetics) and inhibitors of DPP-4 activity (incretin enhancers) have been found to be promising in lowering the glucose level in the experimental as well as clinical studies [54-57]. Interestingly, GLP-1 expression has been found in the kidney of rats and the rGLP-1 found to inhibit the Na+ reabsorption in the proximal tubule and increases glomerular filtration rate in kidneys [16,17]. In addition, GLP-1 has been noted to improve endothelial dysfunction in type 2 diabetic patients with coronary heart disease [18]. Moreover, treatment with exendin-4 (GLP-1 receptor agonists) was found to show the significant reduction in glomerular hypertrophy, mesangial matrix expansion, TGF-β1 expression, and type IV collagen accumulation and associated glomerular lipid accumulation in db/db mice [19]. These studies support the direct renoprotective effect of saxgliptine observed in the present study.
Accumulating research suggests that oxidative stress is a significant contributor to the pathogenesis of diabetic nephropathy. Lipid peroxidation is initiated by free radicals attack of membrane lipids, generating large amounts of reactive products, which have been implicated in pathogenesis of diabetic complications including diabetic nephropathy nephropathy. Thereby, increase in renal TBARS and decrease in GSH levels are considered to be an index of development of oxidative stress [58]. In the present study, diabetes has been noted to increase the renal TBARS and decrease the GSH levels in the kidney. Thus, it may be suggested that diabetes-induced development of oxidative stress may develop nephropathy by damaging renal architecture. In the present study, the administration of either fenofibrate or saxagliptin partially reduced the development of oxidative stress in diabetic rats with nephropathy by reducing renal TBARS and concomitantly elevating renal GSH levels. However, their combination markedly prevented the development of renal oxidative stress in diabetic rats with nephropathy. Thus, it may be suggested that the direct renoprotective potential of combination of fenofibrate and saxagliptin in preventing diabetic nephropathy may be due to their anti-oxidant properties on diabetic kidney. Taken together, the overall observed beneficial effect of combination of fenofibrate and saxagliptin in preventing the development of diabetic nephropathy may be attributed to their direct renoprotective action, improving altered lipid profile and reversing renal oxidative stress. The renoprotective effect of lisinopril has been well reported in basic and clinical studies [59,60]. Therefore, lisinopril has been employed as a standard drug in the present study. The renoprotective effect of combination of fenofibrate and saxagliptin observed in present study was slightly better than lisinopril treatment in ameliorating diabetes-induced nephropathy.
On the basis of above discussion, it may be concluded that the concurrent administration of fenofibrate and saxagliptin may have prevented the development of diabetes-induced nephropathy by reducing the altered lipid profile, decreasing the renal oxidative stress, and providing the direct nephroprotective action. In addition, their combination strategy may provide synergistic renoprotective effect against diabetic nephropathy. Therefore, long-term clinical studies demonstrating the rationale of combination of fenofibrate and saxagliptin in preventing diabetic nephropathy are mandatory.
The authors convey their sincere thanks to Dr. Rani Bansal, H.O.D, Department of Pathology, Subharti Medical College, Meerut, India for her valuable technical support in histological study.
