Research Article - (2013) Volume 2, Issue 2
While the use of sentient animals in biomedical research is known to be ethically contentious, the fact that scientific issues surround the practice is typically ignored. The justification for using sentient animals in research relies on the alleviation of human suffering that purportedly results from such studies and that such breakthroughs cannot occur without studying whole, intact, biological systems. As all biological systems are examples of evolved complex systems, we question the ability of one evolved complex system to predict outcomes for a second. A classic claim regarding the advances in medical care arising from the use of intact sentient animals in research is the discovery and development of antibacterial agents. We give a brief overview of infectious disease research in the 19th century, then examine the history of antibacterials in the first half of the 20th century. We focus on the use of intact sentient animals to test antibacterials for efficacy and side effects-which is also the primary use of animal models in drug development today. As the development of antibacterials is frequently cited as an illustration of the necessity of using intact animal models in drug development, we also examine the impact this position has had on drug development in general. The development of antibacterials, and drugs in general, is placed in the context of evolution, complexity science, and conserved processes. We conclude that the commonly related history revolving around the necessity of intact animal models in the development of antibacterials is not entirely accurate and that this finding has ethical, financial, legal, medical, and scientific ramifications.
Keywords: Animals; Antibacterials; Biological complex systems; Drugs; Epistemology; Ethics; Evolution; History
The fact that the use of intact sentient nonhuman animals (here after referred to as animals) in science and biomedical research is ethically controversial is well known [1-6]. For example, Giles, writing in Nature in 2006, states: “In the contentious world of animal research, one question surfaces time and again: how useful are animal experiments as a way to prepare for trials of medical treatments in humans? The issue is crucial, as public opinion is behind animal research only if it helps develop better drugs. Consequently, scientists defending animal experiments insist they are essential for safe clinical trials, whereas animal-rights activists vehemently maintain that they are useless” [7]. Likewise, an editorial in Nature in 2009 states: “Animal-research policies need to be guided by a moral compass-a concensus of what people find acceptable and unacceptable” [8].
Less appreciated however, is the fact that some uses of animal models have been questioned on scientific grounds [1,9-23].The justification for currently using intact sentient animals revolves around the advances in medical care that have allegedly come from using them as models. One example of such a medical advance is the discovery and development of antibacterial agents, commonly referred to as antibiotics. Gamble summarizes the position we are referring to when she writes: “The case for experimenting on animals in order to advance medicine is that it will produce such great benefits for humanity that it is morally acceptable to harm animals. Throughout the 20th century, research using live animals has led to ground-breaking advances in medicine and treatment for human diseases: such as organ-transplant techniques, antibiotics, modern anaesthetics, the vaccine for Polio and the vital discovery of insulin as a treatment for diabetes . . .”[24].
We will give a few more examples of the above position so the reader does not think we are creating a straw man. The US-based Foundation for Biomedical Research states: “Animal research has played a vital role in virtually every major medical advance of the last century, for both human and animal health. From the discovery of antibiotics . . .”[25]. Similarly, the American Medical Association states: “. . . virtually every advance in medical science in the 20th century, from antibiotics and vaccines to antidepressant drugs and organ transplantation, has been achieved either directly or indirectly through the use of animals in laboratory experiments” [26]. Finally, the UK-based Understanding Animal Research states: “If you have ever had a vaccination, taken a headache pill or been given an antibiotic, you have benefited from animal research” [27].
While we acknowledge that many scientific advances have involved animals, we question the above position and view it as a sweeping generalization. Reality is rarely so straightforward. We will examine the role animal models played in the development of antibacterials in order to ascertain whether perhaps other examples from the above should also be re-examined, and to assess the ramifications of uncritically accepting such claims.
As the field of medicine uses words in ways that may differ from their common or lay usage, as well as differing among specialties, we will take a moment to define some terms relevant to this subject.
• Many people refer to the medications that cancer patients receive as chemotherapy. However, in medical science, chemotherapy initially meant the use of a chemical to combat disease, usually infectious disease. Ehrlich coined the term chemotherapy in the 19th century [28]. Today, the word essentially always refers to drugs used as cancer treatments.
• Antimicrobial drugs are chemicals that kill microbes that are dangerous to the organism that produces the antimicrobial. Antimicrobials theoretically have minimal toxic effects to the organism since they are primarily toxic to the invading microbe.
• Antibiotic is usually used to mean chemicals that kill bacteria. Actually the word, coined by Waksman [29], means a chemical produced by a microbe that is used to kill or inhibit another microbe. Thus, sulfonamides would not be included in this category because they are synthesized.
• Antibacterial is used to refer to chemicals that kill or inhibit bacteria.
• Antibiosis is a term used in the 19th century meaning “against life,” and referred to the properties of early antibacterials [30,31].
• A medium is a solution used to grow bacteria or viruses. The medium can be liquid or solid and different media can be manufactured to grow only certain microorganisms. Minimal medium contains the only minimal requirements for a species to grow.
We surveyed the scientific and medical literature concerning topics relevant to animal models and the development of antibacterials and drugs in general. These topics included:
• Animal models
• Philosophy of science and epistemology
• Evolution
• Drug development
• Biological complex systems
• History of antibacterials and infectious diseases
We sought to place the recorded history of drug development into the background of evolved complex systems. This would allow us to determine the advantages and limits to using intact animals as models for humans in general, as well as the role played by animal models in the development of antibacterials.
Animal models as evolved complex systems
Animals are used in numerous ways in science and research (Table 1). It will be our contention in this article that while animals can be successfully employed in categories 3-9 in Table 1, they are not of predictive value for human response to drugs and disease. The reason for this is that animals and humans are examples of biological complex systems. Specifically, they are evolved complex systems, and one evolved complex system cannot be of predictive value for a second evolved complex system when the analysis involves higher levels of organization. These higher levels of organization are where responses to drugs and disease occur. We begin our analysis with an overview of complexity science.
| 1. Animals are used as predictive models of humans for research into such diseases as cancer and AIDS. |
| 2. Animals are used as predictive models of humans for testing drugs or other chemicals. |
| 3. Animals are used as “spare parts”, such as when a person receives an aortic valve from a pig. |
| 4. Animals are used as bioreactorsor factories, such as for the production of insulin or monoclonal antibodies,or to maintain the supply of a virus. |
| 5. Animals and animal tissues are used to study basic physiological principles. |
| 6. Animals are used in education to educate and train medical students and to teach basic principles of anatomy in high school biology classes. |
| 7. Animals are used as a modality for ideas or as a heuristicdevice, which is a component of basic science research. |
| 8. Animals are used in research designed to benefit other animals of the same species or breed. |
| 9. Animals are used in research in order to gain knowledge for knowledge’s sake. |
Table 1: Nine categories of animal use in science and research [8].
Systems can be variously described. There are open systems and closed systems, physical and abstract systems, subsystems and super systems, natural or manmade systems, among others. Systems can also be described as simple, complicated, complex, or chaotic. We will focus on complex systems but will describe simple systems for the purposes of contrast. A simple system has few components and can usually be understood intuitively. Merely by looking at the system, one can usually understand the components and how they interact. A bicycle would be an example. Simple systems are nothing more than the sum of their parts, are not influenced by their environment and react to perturbations in a linear fashion. A Swiss watch, while complicated, is still a simple system.
Conversely, a complex system interacts with its environment-is dynamic-and adapts to its environment. In the case of living systems, the complex system may adapt by evolving. The complex system is a system of systems. Some of these systems may be simple but others are complex and even chaotic. The number of components in a complex system is usually very large and the components exist across a scale of size. The components can be grouped into modules and there is a hierarchy of organization. The state of a complex system is very dependent on initial conditions and the system contains feedback loops. Importantly, complex systems have emergent properties, which cannot be derived even with total knowledge of the components. Thus, reductionism can be used to study complex systems, but there are aspects of the system that cannot be discovered by use of reductionism [32-57].
Moreover, complex systems respond to perturbations in a nonlinear fashion. Redundancy and robustness are also characteristics of a complex system. One reason a living system is robust-resists change-is because of a redundancy of genes and gene products. For example the same protein may be the product of the exons of one gene or a combination of the exons of two or more genes. The process of adding exons together to produce proteins is called alternative splicing. Lastly, complex systems are non simulable. This is obviously important in terms of modeling [32-57].
Jensen and Arcaute summarize complexity science thusly: “The science of complexity emphasises the interactions between components. It stresses that components, most often, are heterogeneous and evolve in time. Complexity is concerned with the emergent properties at systems level originating from the underlying multitude of microscopic interactions. . . . Complex Systems consist of a large number of interacting components. The interactions give rise to emergent hierarchical structures. The components of the system and properties at systems level typically change with time. A complex system is inherently open and its boundaries often a matter of convention” [58].
Barbosa-Morais, et al. [59] discovered that alternative splicing is important in species evolution. Hochachka and Somero summarize the problem Barbosa-Morais, et al. [59] addressed: “The problem, and in some sense the paradox, is that protein and gene sequences in the common chimpanzee and in humans are remarkably similar. In fact, human and chimpanzee proteins appear to be nearly 99% identical at the amino acid level, and it is widely assumed that the same percentage similarity prevails at the DNA level. Yet no one could mistake the two species as one” [60]. Barbosa-Morais, et al. [59] compared hundreds of thousands of genetic messages in organs from 10 different vertebrate species in order to gain greater appreciation for species differences. They discovered that evolution used alternative splicing and that had a dramatic effect on the structure and complexity of genetic messages. Just as complexity science predicted, small changes in initial conditions, like alternative splicing, can lead to major changes in outcomes to perturbations such as disease and drugs.
Likewise, changes in regulatory genes have also had a major impact on initial conditions. Hochachka and Somero state: “The differential expression and differential evolution of these kinds of genes [regulatory genes] supply raw material for evolutionary change and species specificity . . . the complexity of physiological systems in multicellular organisms requires ever more complex sensing, signal transduction, and communication, as body plans attain higher levels of complexity” [60]. They continue: “What these examples suggest is that only exceedingly minimal changes in genome sequences may be necessary to specify separate species, possibly with larger percentage changes in gene expression patterns. Of course, the longer any two such related lineages evolve separately from each other, the greater the genetic differences between them may become” [59]. Kirschner and Gerhart support this idea, stating: “Third, all organisms are a mixture of conserved and non conserved processes (said otherwise, of unchanging and changing processes) rather than a uniform collection of processes that change equally in the course of evolution. Novelty in the organism’s physiology, anatomy, or behavior arises mostly by the use of conserved processes in new combinations, at different times, and in different places and amounts, rather than by the invention of new processes . . . Most evolutionary change in the metazoa since the Cambrian has come not from changes of the core processes themselves or from new processes, but from regulatory changes affecting the deployment of the core processes” [61].The notion that regulatory genes are responsible for major changes during evolution is now more or less universally accepted [62,63].Gene expression also varies greatly intra- and inter-species, in humans [64-67] and in animals [68-71].
Convergent evolution, the acquisition of the same trait in species with different evolutionary ancestors, is another reason for species variation. The classic example of convergent evolution is the trait of flight in bats, birds, and insects. Even though the trait evolved through different mechanisms, the outcome-the ability to fly-is shared by all. Another example is the eye of cephalopods, for example the octopus, and the eye of vertebrates. This also demonstrates the same outcome, but by very different mechanisms. Other examples include: blonde hair evolved in Melanesians by a different mechanism than it did in Europeans [72];ion selectivity in neuronal signaling channels evolved at least twice in animals [73]; and Odeen and Hastad discovered that birds evolved ultraviolet vision multiple times, all due to single nucleotide polymorphisms [74].
In addition to the above, species differences can arise due to mutations like copy number variants (CNVs) and single nucleotide polymorphisms (SNPs), a difference in the total number of genes, pleiotropy, transposable elements, different genes performing the same function and vice versa, an old gene can be used for a new function, and epigenetics. In the context of a complex system all of these changes are of major importance in terms of predicting human response to drugs and disease.
The above does not derail the use of animals in science, however. Animals can be successfully used in many ways per categories 3-9 in Table 1. Animals have a long history of contributing to science in the form of heuristics: guides to discovery, stimulating interest as a means of furthering investigation. For example, by studying sea sponges scientists created a synthetic hybrid material composed of approximately 90% minerals. Despite its high mineral content, the material is very flexible. Imitating the spicules of the sponge was the key to the strength and flexibility of the new material. The new spicules are also able to transmit light and maintain this property even when they bend [75].
There are some outcomes from perturbations to complex systems that can be predicted by using one complex system to model and predict such outcomes for a second. The key to this, however, is that the perturbation must occur at a lower level of organization. For example, dropping a frog out of an airplane flying at 30,000 feet above ground level will result in massive trauma to the frog; it will do likewise to a human. Perturbations that involve conserved processes are also likely to result in similar outcomes but there are exceptions that limit this use [76]. So, while animals can be used effectively in research and science, there is a limit to using one evolved complex system to substitute for another even when studying conserved processes. Krakauer et al. [77] , state: “Scientific theories seek to provide simple explanations for significant empirical regularities based on fundamental physical and mechanistic constraints. Biological theories have rarely reached a level of generality and predictive power comparable to physical theories. This discrepancy is explained through a combination of frozen accidents, environmental heterogeneity, and widespread non-linearities observed in adaptive processes” [77]. Because evolution imposed changes on complex systems (illustrated in Figure 1 (used by permission of RG)), we will show that animal models are not of predictive value for human response to drugs and disease. We now turn to the antibacterials.

Figure 1: Dynamics of Evolved Complex Adaptive System. Each of the tools below employed by evolution is a component or module in a complex system. The changes caused by evolution will effect the CAS in a nonlinear fasion. The differences among species include all of the below. The thousands of defferences between two species result in a species that is differently complex from its ancestors and cousins.
19th century research on infectious diseases
In order to fully appreciate the discovery and development of antibacterials, we need to start with a brief history of infectious disease research, focusing on the 19th century. The earliest antimicrobial was probably cinchona bark, which was used to treat malaria prior to the 17th century. By the end of the 18th century, Hooke had used a microscope to view what he would call cells. The cells were from a dead cork plant. Over the next several years, Hooke would describe many structures viewed with the microscope. During this time period, van Leeuwenhoek also described the bacteria and protozoa that he saw under the microscope.
The mid to late 1800s saw many advances concerning infectious diseases. By1869, Bechamphad synthesized a chemical compound, Atoxyl, which included arsenic. He realized arsenic was toxic and combined it with the dye aniline. His work was largely ignored. In 1870, Burdon-Sanderson noticed that culture medium that had been contaminated with Penicillium mold did not grow bacteria. Interestingly, he worked at St Mary’s in London where Fleming would rediscover this same action from this mold [[78]as quoted in [79]].In 1872, Ferdinand Julius Cohn published a landmark paper on bacteria and the cycling of elements. He used a classification method that contained the name Bacillus. Brefeld also reported that he had grown fungus on gelatin [80]. In 1874,William Roberts used a liquid broth to grow Penicillium glaucum and observed that the medium resisted bacteria [81]. In 1875, John Tyndall noticed that some fungi inhibited the growth of some bacteria, and Schroeter grewbacterial colonies using slices of potato as the medium.
In 1877, Pasteur and Joubert noticed that, in animals that were co-infected with Bacillus anthracis, anthrax did not develop as well as it did in the other animals [82]. Pasteur observed that: “if we could intervene in the antagonism observed between some bacteria, it would offer perhaps the greatest hopes for therapeutics” [83]. Pseudomonas aeruginosa and Streptococcus erysipelatis were also found to inhibit the growth of certain bacteria [84]. Babès and Garrénoticed that the secretions of certain bacteria could kill other bacteria [85,86]. Emmerich discerned that a guinea pig he had infected with Vibrio cholera did not manifest cholera as expected. He learned that the guinea pig had previously been infected with Streptococcus erysipelatis. He went on to demonstrate that anthrax could be prevented by prior infection with Bacillus anthracis [87].Koch developed methods for staining bacteria, among other things.
Prior to the agar plate methods used today, bacteria were usually grown in animals or in nutrient-rich broths. The broths were meat extracts or serum from cows, although Koch originally tried egg and starches [88]. These broths quickly became the standard for Koch’s lab although there were problems with the method: growing an isolated and pure strain of bacteria was essentially impossible. Koch, like Schroeter, noticed that bacteria would grow in slices of potato when they were left out for periods of time and he took advantage of this when attempting to grow Bacillus anthracis [88]. The potato method did not work as well for other bacteria, however. Koch then tried solidifying the medium by using gelatin. This proved unsatisfactory as the medium would melt on warm days and certain bacteria released the enzyme gelatinase that would digest the medium leaving a dish of goo.
In 1881, scientist Walter Hesse and his wife, Angelina Fannie Eilshemius, who assisted him, joined Koch’s lab. Fannie Eilshemius drew pictures of what Walter saw under the microscope. Walter asked her how she managed to keep the deserts she made from melting during the summer months and she replied that she used agar-agar, a product she had learned about as a child. Agar-agar, a polysaccharide derived from red seaweeds, has long been used for cooking in Asia. Walter discussed this with Koch, who added agar to his nutrient broths and found that the resulting solid culture medium remained solid and allowed growth of the microorganism [80,89,90]. Koch also replaced the usual flat glass plates that contained the broth with a double-sided dish suggested by his associate, Julius Richard Petri. The Petri dish, with agar-based nutrient solution, became the standard used in microbiology [80,89]. This technique allowed Koch to isolate specific bacteria of which the first was . In 1905 he was awarded the Nobel Prize for his work on tuberculosis. Koch also developed methods for obtaining pure cultures of bacteria.
In 1884, building on work that discovered that certain dyes would bind to cells and bacteria, Gram developed a system for identifying bacteria-the Gram stain. Loeffler added peptone and salt to Koch’s meat extract [91] as cited in [92]]. The peptone added amino-nitrogen and the salt raised the osmolarity of the medium. In 1885, Ehrlich described the inhibition of syphilis by arsenic [93].The German rabies vaccine was also developed in 1885. In 1888, Freudenreich observed that the growth of typhoid bacilli was inhibited when he attempted to grow it using broth in which bacteria had previously been grown [94].In 1889, Bouchard infected rabbits with P.aeruginosa resulting in protection against anthrax [95]. Beijerinck described what would become known as agar diffusion. He used it to study how different auxins effected the growth of various bacteria [96].
By the end of the 19th century, Petri dishes with agar, nutrient broth, and peptone were routinely being used. Selective isolation of bacteria was more common, using dyes and other chemicals as demonstrated by Ehrlich. (It would not be until the 1960s that antibacterials would be used in order to inhibit bacterial growth in Petri dishes, thus allowing isolation and growth of select bacteria [97-100].)In 1891, the diphtheria antitoxin was developed and in 1895, Tiberio noted that mold could inhibit the growth of bacteria. He demonstrated that P. glaucum inhibited staphylococci and other bacteria. He tested it on animals and reproduced the findings. Gosio noted similar properties of the mold in 1896 ([101]and [102] as cited in [79]). Martinus Beijerinck had discovered viruses and developed enrichment cultures [103,104].
Many of the above advancements were developed or discovered by using animals. Much was also done using in vitro methods. Serendipity and hard work also figured into the above breakthroughs. An interesting historical and scientific discussion could be undertaken regarding the breakthroughs cited above in terms of which were dependent upon using intact whole animals and or the products from animals. Could these breakthroughs have been accomplished without such uses? While such a discussion would no doubt be stimulating, such is not our interest here. First, there is little such a discussion could contribute to the ethical debate concerning the use of animals in science and biomedical research as society long ago accepted the use of important medical information obtained even in the most unethical of fashions. The use of information from Nazi concentration camps and other Nazi-conducted experiments of that era, as well as the data from Japanese Unit 731, are cases in point. Second, scientifically evaluating the role animals played in the above breakthroughs would require detailed technical knowledge from a variety of fields. Such are the things master’s theses are made from. Regardless of how detailed such an examination would be, the fact would remain that the use of animals in research today is scientifically very different from that of the 1800s. Third, the subject of this article is the development and discovery of antibacterials as occurred in the 20th century. We now turn to that subject.
Salvarsan
Salvarsan, meaning saving arsenic, also known as arsphenamine and dioxydiamido-arseno-benzol dihydrochloride, is an arsenic-based chemical and is considered the first chemotherapy. In the early 1900s, Schild and Wolferstan revived Béchamp’s arsenic compound and used it to treat skin infections [105] and sleeping sickness from trypanosomiasis. Arsenic compounds were already being used to treat equine and bovine trypanosomiasis but toxicity was a known problem. Wolferstan tried the compounds on rodents, monkeys, and rabbits. This research eventually led to the discovery of salvarsan by Ehrlich and Hata.
Ehrlich and Hata discovered salvarsan in 1909. It was 6th chemical tested in the 6th group of 100chemicalsbeing tested. The total number of compounds having been tested was 606, so salvarsan is frequently referred to as 606. Ehrlich and Hata were seeking a “magic bullet” to kill microorganisms, and since the toxic effects of arsenic were well known, they built their efforts on the element. Mercury was already being used to treat syphilis, caused by the spirochete Treponema pallidum, but there were toxicity problems thus a new drug was needed. The Wasserman blood test, developed in 1906, allowed definitive diagnosis of syphilis and confirmation of cure. Salvarsan was tested on a rabbit infected with Treponema pallidum and shown to be effective and without side effects. Human trials quickly followed [106]. However, toxicities like seizures, renal failure, optic neuritis, and fevers did, in fact, develop in humans. There were also deaths. The side effects, combined with the problems with preparing salvarsan, resulted in it not being as successful as originally hoped [107,108]. Eventually, other animals were tested or treated with salvarsan and toxicities observed in some species [108]. Neosalvarsan (neoarsphenamine), compound 914, developed and marketed in 1912, had fewer problems and replaced salvarsan. Nevertheless, when penicillin became available, the arsenic-based compounds were discarded because of the toxicity problems. Ehrlich was awarded the Nobel Prize in 1908 for his work on immunity. Interestingly, the mechanism of action of salvarsan is still unknown [109].
Salvarsan was shown to kill bacteria and other microbes in animals. This will be a theme throughout this article: animal models can be used to demonstrate antibacterial activity. The relevant fact, in terms of our complex systems analysis above, is that the organism being affected by the antibacterials is not the human but the bacterium. The side effects from the antibacterial, however, affect the human. With few exceptions, efficacy of antibacterials can currently be demonstrated in vitro. Thus, the reason antibacterials appear to be exceptions to the complex systems rule, is that the organism being affected is not the human but the bacterium. When the organism in question is the human, as is the case for side effects from antibacterials, then the animal model fails, as we will see.
One exception continues to be T. pallidum, which cannot be reliably grown in culture medium to this day. There have been various partial successes over time but none reliable enough to employ routinely. T. pallidum obtained from humans was apparently grown in culture by 1906 but the technique was not perfect and then, as now, there were problems with virulence [108,110]. In 1919, scientists described salvarsan as being studied in vitro for its ability to kill parasites, describing it as a “powerful trypanocide and a feeble bactericide,” and noted that spirochetes and trypanosomes seem to react similarly to chemotherapies [111]. In vitro study of spirochetes was available in the era of salvarsan development and such studies did demonstrate that salvarsan was spirocheticidal [112-116].However, Ehrlich stated that salvarsan did not kill spirochetes in vitro [117].In light of other studies that claim salvarsan did kill spirochetes, we assume there were technical issues involved then, as there frequently are now among labs. With the sequencing of the T. pallidum genome [118], there is optimism for further research despite the inability to grow the microbe in culture.
Relevant to this discussion, some animals were more sensitive than others to toxicities; the rabbit originally tested apparently gave no indications of the toxicity problems. Thus, testing 606 on rabbits did not yield reliable information regarding side effects in humans. It appears that in vitro means of determining whether salvarsan was spirocheticidal were available around the time of development but rabbits appear to have been the primary growth medium used and tested. Based on the above, it is difficult to claim that animals were necessary for the development of salvarsan, although they certainly were extensively used.
Prontosil
The history of sulfa drugs dates back to the dye industry where it was discovered that certain dyes bound to cells and or were able to stain bacteria. The azo dyes were known to have antiseptic properties ([119] and [120] as quoted in [79]). This was the basis for the research into ascertaining whether they could be developed into antibacterials. Sulfonamidochrysoidine, also called KI-730 and Prontosil, was synthesized in 1932,by Klarer and Mietzsch who were working for Bayer [121]. Domagk, employed by the chemical and dye company I.G. Farbenindustrie, tested the sulfa-based chemical for antibacterial properties in animals and humans [122] and found that it was effective against hemolytic streptococcus infections such as Streptococcus pyogenes. Prontosil and its derivatives werenot effective in vitro, due to the fact that the active ingredients was dependent upon metabolism, but were effective in animals such as mice. Moreover, it was effective against certain streptococcal strains, but not the ones that affected humans [123,124].The preclinical results from other countries were also confusing but animal studies were eventually performed that resulted in very impressive data. Finally, the drug was administered to humans, including some suffering from puerperal infection, and shown to be very effective [125].The active portion of the compound turned out to be sulfanilamide, which had been used in the dye industry for years, hence was not eligible for a patent. Later sulfa drugs were shown to be effective against streptococcus in vitro, as the active ingredient was being tested. This inspired the development of more sulfa-based drugs [126,127].Domagk was awarded the Nobel Prize in 1939 for his work on the sulfa drugs.
The side effects of the sulfa drugs are minimal in most cases and were not cause for concern. Very little is even written about side effects in the early days of the administration of the sulfa drugs. The antiseptic effects of the azo dyes from which the sulfa drugs were developed were known prior to their development as antibacterials. Some studies on animals were very impressive in demonstrating that the animals treated with the sulfa drugs were not killed by bacterial infections when all the other animals so infected were killed. This helped to convince scientists that the sulfa drugs were worth pursuing. In retrospect however, the efficacy of the sulfa drugs vis-à-vis the impressive results from administration to animals, was to be suspected based on the known properties of the azo dyes. The unknowns concerning the sulfa drugs were side effects and the pharmacokinetic properties, neither of which can be ascertained from animal studies.
Penicillin
The interlude between the sulfa drugs and penicillin saw the discovery ofmore antibacterials that were effective in animals but that could not be administered to humans for various reasons. Tyrothrycin, produced by Bacillus breves, was discovered in 1939 [128-130] in an attempt to find a chemical active against pyogeniccocci. It was shown to be effective in mice but proved too toxic for humans. Gramicidin, also discovered by Dubos [131-134],was similarly shown to protect mice against infections with type III pneumococci but was toxic in humans [134].Both drugs could be applied topically, however.
The biggest breakthrough in antibacterials was penicillin. Fleming actually rediscovered penicillin. Steffee states that: “Folklore of the mid-19th century encouraged the application of mold to a fresh wound as protection against subsequent infection” [135]. As we discussed above, Burden-Sanderson and others had noted that Penicillium mold inhibited the growth of bacteria [81,135-137]. Fleming tested penicillin in vitro and in vivo. He mentions rabbits in his original paper. The in vitro results revealed bacterial inhibition and topical application on rabbits was effective. There was no doubt about penicillin’s antibacterial property. In an attempt to ascertain the effectiveness of penicillin systemically, Fleming tested it in rabbits. Penicillin was eliminated, in the urine, very rapidly inrabbits leading Fleming to believe it would be ineffective for humans with systemic infections. Based on this animal model, he turned his attention to other projects. In reality, the very low concentration of the penicillin injection was responsible for rabbit data, not a species difference.
Fleming did however administer penicillin topically to colleagues and others [136,138-141], including four patients suffering from ophthalmic neonatorum. Three were cured [136,142,143]. Others also administered penicillin to patients during this time [136,143-147]. Despite the fact all these were topical administrations, they did encourage Florey to continue his work on purifying penicillin. Henderson states that Florey: “recalled Paine’s . . . successful topical treatment of ophthalmic neonatorium with a crude broth of penicillin” and that this gave Florey hope that a purified penicillin might be effective systemically [142].
Florey and Chain eventually did produce a purer penicillin than was previously available-the penicillin they produced was shown to have a strength of 5 U/mg whereas later preparations were around 1800 U/mg [148]. Subsequently, they were encouraged by the results of administering their product to infected mice. The claims regarding animal models and the efficacy of penicillin largely center on the use of mice in the development of penicillin. Florey and Chain tested penicillin on mice showing that mice treated with penicillin did not die from infection as all the other mice did. The results from the mouse studies led to a worldwide effort to develop penicillin. But if we are to give animal models credit for this outcome then we must also debit animal models (Fleming’s rabbit) for the long delay. Moreover, as we have pointed out, the antibacterial properties of penicillin were not in doubt. In vitro, in vivo animal studies, and human administration proved it was antibacterial. The question revolved around what penicillin would do to an intact system. As we will discuss momentarily, and as we have pointed out previously [9,11,12], the response of an intact system to a drug varies greatly depending on the species and strain of the intact system. Florey, et al. [143,150], were just as mistaken in believing that what happened in mice would be reproduced in humans as Fleming was in thinking the same about rabbits and humans.
A natural question arising at this time is why didn’t Fleming test what would turn out to be a wonder drug on humans. Weisse sates that Fleming was discouraged about penicillin’s possible use for many reasons including: “…after injection into an ear vein of a rabbit and with blood samples taken periodically thereafter for testing, it was found that penicillin was rapidly removed from the bloodstream. Samples taken at 30 minutes were found almost completely devoid of activity. Of what use might be an antibacterial agent that took several hours to act but was removed from the body within 30 minutes and inhibited by the blood with which it would obviously be mixing” ?[148].
Steffee reinforces this discouragement, stating: “Fleming considered penicillin a potential chemotherapeutic agent, but his early in-vivo investigations were discouraging. In rabbits, serum levels of penicillin dropped rapidly after parenteral administration, too fast to allow the several hours of contact with bacteria required for an effect in vitro”[135].
When evaluating the role of animal models in the development of antibacterials, one must consider the misses as well as the hits. Penicillin began as a huge miss. Fleming very clearly equated response in humans with response in rabbits and abandoned one of the most life-saving drugs of all time on this basis. Steffee defends Fleming’s abandonment of penicillin stating: “. . . how many therapeutic modalities with the poor in vivo results of Fleming’s early penicillin trials would be offered continued funding today [135]?” Weisse also defends Fleming: “One might well wonder why, given the uncontrolled devastation of bacterial diseases, no further experiments on animals or humans were undertaken. The rapid disappearance from the blood has already been mentioned . . . Even the choice not to use animal experiments more extensively, a routine practice of investigators on the continent, could be defended by Fleming and his group. After all, there might be differences between humans and other animals in resistance or susceptibility to different infections”[148].
Fleming noted the differences in species’ responses to penicillin when he stated: “How fortunate we didn’t have these animal tests in the 1940’s [sic], for penicillin would probably never been granted a license, and possibly the whole field of antibiotics might never have been realized” (as quoted in [149]). Florey did the same when he stated: “Mice were used in the initial toxicity tests [by Florey and Chain] because of their small size, but what a lucky chance it was, for in this respect man is like the mouse and not the guinea-pig. If we had used guinea-pigs exclusively we should have said that penicillin was toxic, and we probably should not have proceeded to try and overcome the difficulties of producing the substance for trial in man”[150]. These statement were reinforced by Koppanyi and Avery [151].
Fleming did eventually administer penicillin to a friend who was dying in the hospital from streptococcal meningitis. Many advances in medicine have likewise been secondary to having no other options. This was in 1942 after and the mouse experiments of 1940. Florey gave Fleming his supply of the purified penicillin for use in his friend [148]. The friend improved dramatically after administration. Interestingly, Florey administered penicillin to a cat at the same time Fleming was administering it to his friend. Florey’s cat died [152]. There are actually numerous specie-specific responses to penicillin. Under certain circumstances, penicillin kills guinea pigs and Syrian hamsters [153,154]. In addition, penicillin is teratogenic in rats, causing limb malformations in offspring. This is one of the problems with using animal models to predict human response: Interspecies variation is to be expected when studying evolved complex systems.
Fleming also rediscovered agar diffusion, a method still used to test antibacterials. Aminov writes: “Fleming’s screening method using inhibition zones in lawns of pathogenic bacteria on the surface of agar-medium plates required much less resources than any testing in animal disease models and thus became widely used in mass screenings for antibiotic producing microorganisms by many researchers in academia and industry. . .”[109].This screening method essentially eliminated the need to test drugs for antibacterial properties on animals.
Streptomycin
Selman Waksman, a soil biologist, was awarded the Nobel Prize in 1952 for his discovery of streptomycin in soil samples. Waksman was actually co-discoverer with his graduate student Albert Schatz, who was recognized as an equal partner via the patent for streptomycin [155-158]. Waksman and Schatz were systematically looking for antibacterials effective against gram-negative bacilli because penicillin was effective only against gram-positive. Streptomycin was the first drug effective against tuberculosis (TB) and TB meningitis. It was also effective against Haemophilus influenzae. Waksman used culture medium and the well-plate method to test for antibacterial activity. Waksman, along with another graduate student, Woodruff, had already isolated actinomycin and streptothricin [159-161], but these drugs, along with clavacin, proved toxic.
Schatz used two sources for his Streptomyces griseus. Each was a different strain and both produced streptomycin that he then cultured. One came from the throat of a chicken via Doris Jones’ lab, and the other from a soil sample. Schatz used the one that came from the soil sample preferentially as it produced more streptomycin [157,162-164].Streptomycin was tested by Waksman and others and shown to kill TB in vitro [165-167]. Still, some thought it was merely bacteriostatic [168]. Regardless of the early questions about mechanism [168], it was shown to be effective for treating TB. Resistance proved to be a problem and, although this was not initially seen in animals, it was demonstrated in vitro [167].
Merck had performed the animal and clinical studies of earlier antibacterials isolated by Waksman but streptomycin was evaluated at the Mayo Clinic. Guinea pigs that had been infected with TB were administered streptomycin and demonstrated full recovery [162]. Clinical trials began in 1944. Ototoxicity and renal toxicity proved to be problems in humans, although neither was previously seen in animal studies [169,170]. Otherwise, streptomycin has few severe side effects. In summary, the toxicity of streptomycin was not seen in animals prior to release and efficacy was demonstrated in vitro.
Current practices in drug development
We will now attempt to place the above examples into the context of current knowledge about drug development. This is not meant to condemn the past based on information available only in the present. Rather, we want to discuss what could reasonably be expected of antibacterial development, in terms of safety and efficacy, even using current science. We will examine drug development in general then briefly present an example of an antibacterial.
First, we must explain how the term predict is used in science. We are addressing the predictive value of animal models for human response to drugs and disease. Predictive value can be calculated and is, at least in many situations, objective (Table 2). This use of predict differs from predicting outcomes based on a new hypothesis. Scientists frequently propose a hypothesis, then test it in order to compare the outcome in reality and the expected outcome if the hypothesis is true. This is fundamental science and occurs daily in universities and research institutions. Predictive value has nothing in common with testing hypotheses. People wish to know the predictive value of many things including lab tests, pencil and paper tests, and practices such as the use of drug sniffing dogs to detect drugs. Predictive value can also be calculated for animal models in terms of how well they predict human toxicity, bioavailability, and cancer among myriad other things. These calculations have been performed for numerous types of animal models and the results are clear: animal models have no predictive value. Animal models do have very low predictive value for human response to drugs in terms of efficacy [171-188],or the five properties of absorption (A) [189-194], distribution (D) [195,196], metabolism (M) [197-207], elimination (E) [208,209], and toxicity (T) [194, 210-221], usually referred to collectively as ADMET. Even with current knowledge of physiology and inter-species differences, scientists are unable to predict, to the standards required in medicine, essentially any of the above six properties. Medical science needs predictive values greater than or in the range of 0.9 and animal models usually give positive and negative predictive values in the range of 0.0-0.5 [222-228]. This is what would be expected from chance alone. In many cases, calculations are not necessary as none of the drugs that were effective in animal models, for example neuroprotectants and a vaccine for HIV, were effective in humans. In light of the fact that individual humans respond differently to drugs, an observation that has led to the current emphasis on personalized medicine, it should come as no surprise that inter-species extrapolation fails [229]. Consider the following.
| Gold Standard | |||
|---|---|---|---|
| GS+ | GS- | ||
| Test | T+ | TP | FP |
| T- | FN | TN | |
| T+ = Test positive T- = Test negative T = True F = False P = Positive N = Negative GS+ = Gold standard positive GS- = Gold standard negative |
|||
| Sensitivity = TP/(TP+FN) Specificity = TN/(FP+TN) Positive Predictive Value = TP/(TP+FP) Negative Predictive Value = TN/(FN+TN) |
|||
Table 2: Binomial table for calculating predictive value.
Toxicity derails many drugs in clinical trials and many drugs being marketed have toxicity or other problems. Cross, et al. [230]discovered that the dosage for one in five new molecular entities (NMEs) released in the US between 1980 and 1999 was changed after release onto the market, and that of these, four out of five were decreased for safety reasons. Heerdink et al. [231] found similar changes for drugs released in Europe. Many studies have revealed unanticipated toxicities after a drug has been released to the market [232].
It is estimated that 80% of all drugs that fail in development fail due to problems in ADMET and that 50% of those that make it to market have problems associated with ADMET [233]. Patterson likens the process of drug development to making airplanes and marketing the ones that do not crash[233], not a very flattering portrayal of current drug development. This must be considered in light of the fact that animal models figure prominently in this process. Many studies have noted the inability of animal models to predict human efficacy, which is usually based on animal models, or ADMET [11,12,177,189-191,193,195,196,200,203,207,218-221,233-238].
This failure is acknowledged by industry. An editorial in Nature Reviews Drug Discovery in 2005 states: “Clearly, one part of the problem [of drug research] is poorly predictive animal models . . .” [239]. Littman and Williams of Pfizer acknowledge: “In the new paradigm, studies in humans increase confidence in the relevance of novel drug targets and largely replace the animal efficacy models that are often poorly predictive of the efficacy of novel agents with unprecedented mechanisms of action . . .” [235]. Another editorial in Nature Reviews Drug Discovery in 2011 states: “Unpredicted drug toxicities remain a leading cause of attrition in clinical trials and are a major complication of drug therapy” [217]. Oliff, former executive director for cancer research at Merck explains: “The fundamental problem in drug discovery for cancer is that the model systems are not predictive at all”[174]. Chabner and Roberts note that: “Fewer than 10% of new drugs entering clinical trials in the period from 1970 to 1990 achieved FDA approval for marketing, and animal models seemed unreliable in predicting clinical success . . .” [240].
Björquist, et al. [176]., state that: “Furthermore, the compound attrition rate is negatively affected by the inability to predict toxicity and efficacy in humans. These shortcomings are in turn caused by the use of experimental pre-clinical model systems that have a limited human clinical relevance . . .” [176]. Then-U.S. Secretary of Health and Human Services Mike Leavitt admitted: “Currently, nine out of ten experimental drugs fail in clinical studies because we cannot accurately predict how they will behave in people based on laboratory and animal studies” [173]. Dixit and Boelsterli state: “Traditional animal toxicology tests predict in the range of less than 10% to -70% of all human adverse effects . . .” [210]. Zielinska, discussing animal models of cancer, states: “Mouse models that use transplants of human cancer have not had a great track record of predicting human responses to treatment in the clinic. It’s been estimated that cancer drugs that enter clinical testing have a 95 percent rate of failing to make it to market, in comparison to the 89 percent failure rate for all therapies . . . Indeed, ‘we had loads of models that were not predictive, that were [in fact] seriously misleading,’ says NCI’s Marks, also head of the Mouse Models of Human Cancers Consortium . . .”[241].The above citations are merely a small sample of the numerous works that have expressed the shortcomings of animals as predictive models for humans.
Another question relevant to this discussion is: How many useful drugs have been discarded because of toxicity or lack of efficacy in animals? Artemisinin is remarkably toxic in animals but not so in humans [242-244]. Diclofenac is likewise toxic in animals but not so much in humans [242,245]. Acetaminophen “increases the incidence of induced renal adenomas in rodents” [246]. Metronidazole is effective against Helicobacter pylori in humans, but increases the “incidence of induced colon cancer in rats” [246].
Eason et al. [214], discuss examples of drugs-carbenoxolone, FPL 52757, and amrinone-that were toxic in humans but not the animal models they were tested on, and drugs that were not toxic to humans but were to animal models-ciprofibrate and omeprazole among others. They state: “Preclinical animal toxicity studies are often limited in their predictive value of safety in man, despite the use of metabolism and pharmacokinetic studies to optimize the choice of species, dose, and method of administration” [214]. The problem is simply inter-species variation. For example, chloramphenicol may cause aplastic anemia in humans, dogs do well with it, but cats die from it [247-249]; moreover, it increases lymphomas in mice but not humans [246].
Lazzarini, et al. [250]. state that the: “predictive value of animal studies about osteomyelitis is still unclear” [250]. They point out that serum levels, the ability of the antibacterial to penetrate bone and the pharmacokinetics (what the body does to the drug) may differ among species. Moreover, the dosages and dose intervals used in humans and animal varies in a clinically significant fashion. Of concern is their statement that: “By the time that a particular antimicrobial agent is slated for animal model studies for efficacy in curing osteomyelitis, toxicity studies and pharmacokinetic studies have been performed in animals and humans.” They also state that: “Teicoplanin and linezolid were successful in the treatment of osteomyelitis in clinical trials, despite being completely inactive in two animal model studies of staphylococcal osteomyelitis. Therefore, the value of animal models as predictors of failure should also be carefully assessed”[250].All of the above can be explained by the fact that animals and humans are examples of living complex systems that have different evolutionary histories. Kehrer states: “Small differences in gene structure can make large differences in function”[251].
Ehrlich was looking for a magic bullet and many scientists still are. In light of the fact that humans and animals are evolved complex systems it should be clear that the magic bullet concept is outdated. A drug may be effective in one individual but not another. Likewise, a drug maybe safe for one species but not another. Narayan, et al. [252], state: “The time has come to move beyond product-focused ‘magic bullet’ therapeutic development strategies towards models that can also incorporate devices, tools and services to provide integrated health-care solutions. . . . Part of the reason for this lack of progress is likely to have been too great a focus on developing single ‘magic bullet’ drugs for very complex diseases” [252].
Narayan et al. [252], continue, discussing why magic bullets do not exist for mental illness: “Although serious mental illnesses are among the most heritable of diseases, indicating a strong biological basis, they arise out of the inheritance of multiple susceptibility and protective genes that interact stochastically and with environmental factors to produce phenotypes that manifest as a constellation of symptoms, including changes in cognition, mood, perception and dysregulation of autonomic, endocrine and circadian pathways. It is therefore unlikely that single-target drugs will adequately treat all facets of such complex diseases” [252].
Finally, all of the above must be placed in the context of personalized medicine. Physicians and researchers currently acknowledge that individual humans react differently to drugs and disease [253-257]; even monozygotic twins vary in response [258-270]. Based on this, disease and drug responses are being classified and approached differently from the past (Figure 2) [271]. The goal of personalized medicine is to match gene to disease or gene to drug and administer drugs that are both efficacious and side effect free. This is already happening for many diseases (Table 3) [272]. Since humans vary intra-species in terms of disease and drugs, it is highly improbable that inter-species extrapolation will yield results of predictive value [229].
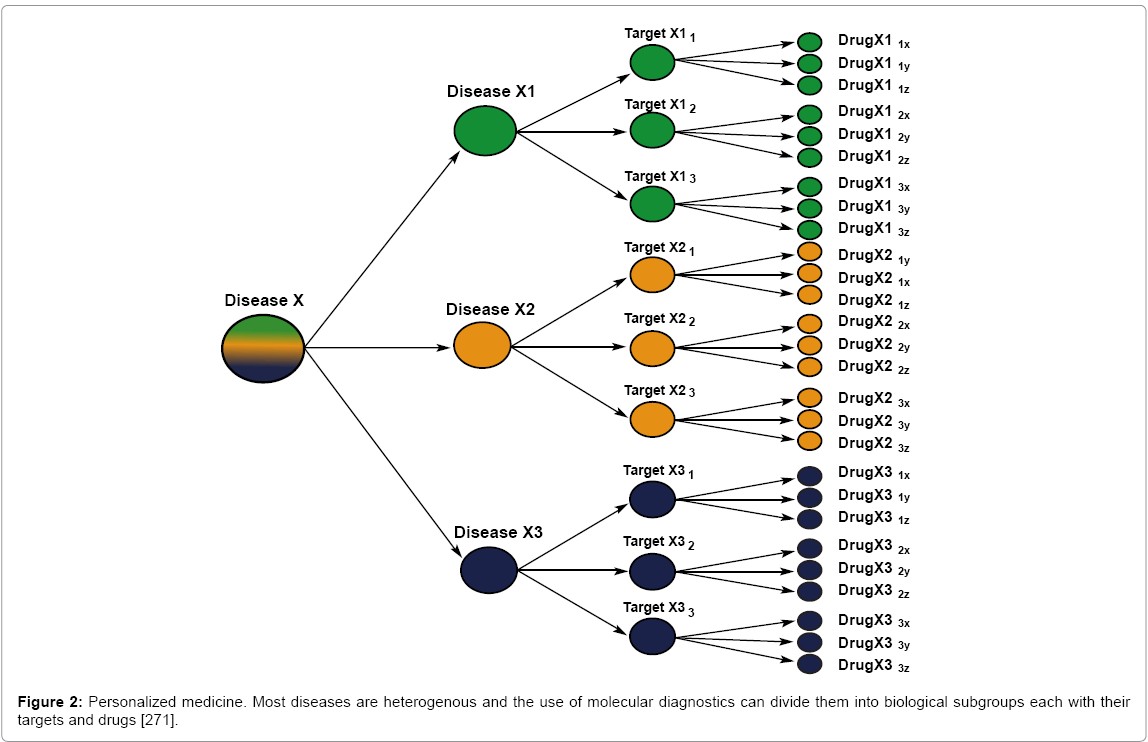
Figure 2: Personalized medicine. Most diseases are heterogenous and the use of molecular diagnostics can divide them into biological subgroups each with their targets and drugs [271].
| Organ or System involved | Associated gene/allele | Drug/drug response phenotype |
|---|---|---|
| Blood | ||
| Red Blood Cells | G6PD | Primaquine and others |
| Neutrophills | TPMT*2 | Azathioprine/6MP-induced neutropenia |
| UGT1A1*28 | Irintotecan-induced neutropenia | |
| Platelets | CYP2C19*2 | Stent thrombosis |
| Coagulation | CYP2C9*2, *3, VKORC1 | Warfarin dose- requirement |
| Brain and peripheral nervous system | ||
| CNS depression | CYP2D6*N | Codeine-related sedation and respiratory depresiion |
| Anaesthesia | Butyrylcholinesterase | Prolonged apnoea |
| Peripheral nerves | NAT-2 | Is oniazid-induced peripheral neuropathy |
| Drug hypersensitivity | ||
| HLA-B*5701 | Abacavir hypersensitivity | |
| HLA-B*1502 | Carbamazepine-induced Stevens Johnson syndrome | |
| HLA-B*3101 | Carbamazepine-induced hypersensitivity in Caucasians and Japanese | |
| HLA-B*5801 | Allopurinol-induced serious cutaneous reactions | |
| Drug-induced liver injury | ||
| HLA-B*5701 | Flucloxacillin | |
| HLA-DRB1*1501-DQB1*0602 | Co-amoxiclav | |
| HLA-DRB1*1501-DQB1*0602 | Lumiracoxib | |
| HLA-DRB1*07-DQA1*02 | Ximelagatran | |
| HLA-DQA1*0201 | Lapatinib | |
| Infection | ||
| HIV-1infection | CCR5 | Maraviroc efficacy |
| Hepatitis C infection | IL28B | Interferon-alpha efficacy |
| Malignancy | ||
| Breast Cancer | CYP2D6 | Response to tamoxifen |
| Chronic myeloid leukaemia | BCR-ABL | Imatinib and other tyrosine kinase inhibitors |
| Colon cancer | KRAS | Cetuximab efficacy |
| GI stromal tumours | c-kit | Imatinib efficacy |
| Lung Cancer | EGFR | Gefitinib efficacy |
| EML4-ALK | Crizotinib efficacy | |
| Malignant melanoma | BRAF V6OOE | Vemurafenib efficacy |
| Muscle | ||
| General anaesthetics | Ryanodine receptor | Malignant hyperthermia |
| Statins | SLCO1B1 | Myopathy rhabdomyolysis |
Table 3: The most significant genetic predictors of drug response [272].
Animals have been successfully used as heuristics, bioreactors, and as a source for serum and nutrients for growth cultures, especially in the 19th century. In the 19th century, animals were also used to demonstrate the various aspects of the Germ Theory of Disease and various physiological properties common to mammals. This was important for the eventual development of antibacterials. Animals were also used as models to assess efficacy and toxicity of newly discovered antibacterials. This is what is usually meant when people state that animals were used for the discovery and development of antibiotics. However, then, as now, toxicity could not be assessed using animals and efficacy could be assessed without animal models, especially after salvarsan. Animals were therefore not necessary for the development of antibacterials as a class even when considered in light of the technology and knowledge available at that time. Animal models cannot predict human response in terms of efficacy and ADMET now and they could not do so then. Antibacterials were an exception, as the organism being acted on, in terms of efficacy, was the bacterium, not the human. Animal models seem to have given the scientists involved in antibacterial development the confidence they needed, in terms of safety, in order to give the drugs to humans. In light of current knowledge, this confidence was unwarranted.
The fact that animal models have no predictive value for safety and efficacy in drug development has ethical implications as, per Giles, society condones animal experimentation only if it improves drug safety and efficacy. There are also financial implications in the form of animal vendors who will no longer profit from the sale of animals to laboratories and researchers who use animals based on their having predictive value. The legal requirement that new drugs undergo testing on animals is superfluous if the testing has no predictive value. Moreover, as animal use in research today is predicated, in part, on the assumption that antibacterial development was dependent upon animal models for safety and efficacy, asset allocation should change and more attention should be paid to the fact that humans are evolved complex systems with all that such conditions imply.
Animals do have value in medicine and science, but not as predictive models. As per Table 1, animals can still be successfully used as bioreactors, a source for replacement parts, basic research, as well as in other areas. But the notion that animals have predictive value for drug efficacy and safety based on the development of antibacterials is not supported by an informed reading of history, nor is it supported by current drug development.
