Research Article - (2017) Volume 5, Issue 6
Phytochemical studies on Dactyladenia floribunda Welw furnished twelve known compounds including (1) tetracosanoic acid, (2) β-sitosterol, (3) lupeol, (4) betulinol, (5) betulinic acid, (6) hennadiol, (7) ursolic acid, (8) oleanolic acid, (9) β-sitosterol-3-O-β-D-glucoside, (10) hexane-1,2,3,4,5,6-hexol, (11) 4’-methoxy-5,6,7- trihydroxyflavone and (12) 5,7,4’,5’-tetrahydroxyflavonol. (6) Hennadiol was subjected to microwave assisted diacetylation and hydrogenolysis. The crude extracts and some of these compounds were tested for their antioxidant and enzyme inhibitory activities.
Keywords: Dactyladenia floribunda ; Diacetylation; Hydrogenolysis; Antioxidant activities; enzyme inhibitory activities
In the course of our investigation of Cameroonian medicinal plants to discover bioactive compound, we have carried out phytochemical studies on Dactyladenia floribunda Welw; which is a small tree belonging to the family Chrysobalanaceae. This family comprises 18 genera and about 533 species represented by trees and shrubs growing in tropical and subtropical lowlands [1]. The investigations on certain genera of this family such as Parinari, Licania, Chrysobalanus resulted on the isolation and characterization of flavonoids, triterpenoids, steroids, and tannins [2-4]. However, no phytochemical or pharmacological studies have so far been carried out on any species of the genus Dactyladenia . In this paper, we report the isolation and identification of known compounds including fatty acid, steroids, triterpenes, sugar and flavonoids from the stems barks of the title plant. The chemical transformations were also carried out on one of the isolates compound namely (6) hennadiol. The antioxidant, lipoxygenase and urease inhibitory activities of the crude extract as well as isolated and synthesized compounds have been determined.
General
Melting points were determined on a Buchi M-560 melting point apparatus and are uncorrected. UV spectra were recorded on Evolution 300 BB, IR spectra were recorded on SHIMADZU, FTIR-8900 infrared spectrometer. Hydrogenolysis reaction were done in 50 mL RB flack with magnetic stirrer (MINI MAG STIRRER DBK-5058/7) and microwave radiation was applied using a domestic microwave oven (Kenstar Model MWO-9808). The 1H,13C and 2D NMR spectra were measured on at 500 MHz Bruker DRX spectrometer. The chemical shifts are expressed in ppm units relative to TMS. The EIMS were recorded on a JEOL MS 600-I mass spectrometer and ESIMS on a QSTARXL mass spectrometer. Silica GF254 plates (20 × 20 cm; 0.2 mm, E. Merck) were used for TLC. Column chromatographic purification was carried out over silica gel (Kieselgel 60; 230-300 mesh). All the solvents were distilled before use. The compounds were detected using an UV-lamp and by spraying with ceric acid solution followed by heating.
Plant material
The fresh parts (blossom, leaves and stem barks) of Dactyladenia floribunda Welw, were collected in April 2014 from Djeufam-Bazou, locality in the West region of the Republic of Cameroon. Plant material was authenticated by Mr NANA Victor, Botanist at the National Herbarium of Yaounde-Cameroon, where a voucher specimen (21720 SRF/CAM) has been deposited.
Extraction and isolation
The air-dried plant material (3.95 kg) was ground, powdered macerated and extracted with methanol at room temperature for 72 h and filtered. The filtrate was evaporated under reduced pressure to afford 268.16 g of crude extract. The part of the crude extract (156.24 g) was chromatographed over normal-phase silica gel open column and eluted with hexane followed by mixtures of hexane/EtOAc and EtOAc/MeOH in increasing order of polarity increasing proportions to obtain nine fractions (DF-I to DF-IX) according to their TLC profiles. The main fractions DF-I (70 g), DF-III (7.15 g) and DF-VII (22 g), were re-chromatographed on silica gel column to obtain compounds (1-12).
Chemical transformations of (6) Hennadiol
Mirowave assisted deacetylation: Acetylation play important role in the isolation and structure elucidation of terpernoids. Acetylation using pyridine and acetic anhydride has been conventionally used with satisfactory results. These but often suffer from the defect of long reaction time period and poor yields. We have used a method using microwave radiation which reduces the reaction time period to seconds.
Microwave is now being used extensively to bring about quicker result in organic reactions [5,6]. Most of the reactions reported so far however involve relatively simple molecules. Our studies reveal that it is equally applicable to complex molecules like triterpenes. Microwave assisted acetylation of (6) Hennadiol are reported herein. No structural breakdown has been observed in any intake.
Hennadiol (6) (60 mg) was dissolved in pyridine (2 mL) and acetic anhydride (0.4 mL) was added. Microwave radiation at full intensity was applied for 120 sec in 30 sec intervals (Figure 1). After every 30 sec a small quantity of the reaction mixture was removed and analysed by TLC. The spot corresponding to compound (6) disappeared completely between 90 and 120 sec. The reaction mixture was cooled and worked up by the usual procedure to yield 3,30-diacetoxy-lup-20(29)-ene (13) (56 mg) as white amorphous powder. Its structure was identified by mass and NMR spectral data.
Hydrogenolysis: Hennadiol (6) (50 mg) was dissolved in CH2Cl2 (5 mL) and MeOH (2 mL). After 15 min of agitation, 5% palladium charcoal (5 mg) was added and hydrogen was passed under stirring at room temperature for 44 h. The progress of the reaction was monitored by TLC every 5 h. After 44 hours, the mixture was filtered and the residue was washed four times with 10 mL of CH2Cl2: MeOH (9:1). The TLC analysis indicated the presence of three compounds which were separated through column chromatography over silica gel eluting with hexane: ethyl acetate (99: 1) to obtain lupeol (3) (17 mg), hexane: ethyl acetate (97.5: 2.5) to obtain 20-3β-hydroxylupan-30-al (14) (10 mg), and hexane: ethyl acetate (24: 1) to obtain 30 norlupan-3β-ol-20- one (15) (12 mg).
DPPH radical scavenging activity
A serial dilution of the test fractions was made in either DMSO or methanol to obtain final concentrations of 500, 250, 150, 125, 62.5, 31.2, 15.6, and 7.8 μM. A 0.3 mM solution of DPPH in ethanol was prepared and 90 μL of this was added to each well of a 96-well plate marked for control and test compounds. Then, 10 μL of each of the concentrations of the compound was added to the particular well to start the reaction. The contents of the wells were mixed for a few sec and then the mixture was incubated for 30 min at 37ºC and the absorbance measured at λ 517 by a micro titre plate reader (Spectra Max plus 384 Molecular devices USA). The assay was standardized using butylated hydroxyanisole (BHA) prior to testing the compounds. Percent radical scavenging activity was determined in comparison with methanol treated control and IC50 was calculated for each compound by EZ fit software (Perrella Software, USA) [7,8].
% DPPH radical scanvenging effect 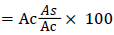
Lipoxygenase inhibiting activity
Lipoxygenase enzyme solution was prepared so that the enzyme concentration in reaction mixture was adjusted to give 130 U per well. 160 μL of 100 mM sodium phosphate buffer (pH 8.0) was added in each well of plate labeled as blank (B substrate and B enzyme ), Control and Test. 10 μL of test compound solution in methanol (of various concentrations 5-500 μM) were added in each well labeled as test. 20 μL Lipoxygenase (20 μL) (LOX) solution was added in each well including B (enzyme ), control, test except B (substrate ). The contents were mixed and incubated for 10 min at 25ºC. substrate solution was flushed with nitrogen gas to avoid auto oxidation before adding to each well. The reaction was then initiated by the addition of 10 μL substrate solution (linoleic acid, 0.5 mM, 0.12 % w/v tween 20 in ratio of 1:2) in each well except B (enzyme ) and the absorbance was measured and recorded at 234 nm. The concentration of the test compound that inhibited lipoxygenase activity by 50 % (IC50) was determined by monitoring the effect of increasing concentrations of these compounds in the assays on the degree of inhibition. The IC50 values were calculated by means of EZ-Fit, enzyme kinetics Program (Perrella Scientific In., Amherset, USA) [5,6].
Urease assay and inhibition
Reaction mixtures comprising 25 μL of enzyme (Jack bean Urease) solution and 55 μL of buffers containing 100 mM urea were incubated with 5 μL of test compounds (1 mM concentration) at 30ºC for 15 min in 96-well plates. Urease activity was determined by measuring ammonia production using the indophenol method as described by Weatherburn. Briefly, 45 μL each of phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) and 70 μL of alkali reagent (0.5% w/v NaOH and 0.1 % active chloride NaOCl) were added to each well. The increasing absorbance at 630 nm was measured after 50 min, using a microplate reader (Molecular Device, USA). All reactions were performed in triplicate in a final volume of 200 μL. The results (change in absorbance per min) were processed by using SoftMax Pro software (Molecular Device, USA). All the assays were performed at pH 8.2 (0.01 M K2HPO4.3H2O, 1 mM EDTA and 0.01 M LiCl2). Percentage inhibitions were calculated from the formula 100-(ODtest well/ODcontrol) × 100. Thiourea was used as the standard inhibitor of urease.
The crude extract (150.16 g) was subjected to column chromatography over silica gel and eluted with hexane followed by mixtures of hexane/EtOAc and EtOAc/MeOH in increasing order of polarity to obtain nine fractions (DF-I to DF-IX). The Fractions DF-I (70 g), was subjected to CC over silica gel (300 g, column: 100 × 8 cm) and eluted with hexane followed by hexane/ EtOAc (1:1) to yield six fractions DFI-a, DFI-b, DFI-c, DFI-d, DFI-e, DFI-f. Compound 1 (20 mg) was obtained from fraction DFI-a, Compound 2 (200 mg) [9], was obtained from fraction DFI-b, Compound 3 (8 mg) and compound 4 (15 mg) [10] was obtained from fraction DFI-d, Compound 5 (40 mg) [11] was obtained from fraction DFI-e and compound 6 (300 mg) [12] was obtained from fraction DFI-f. Fraction DF-III (7.15 g) was subjected to column chromatography (CC) over silica gel (300 g, column: 80 × 4 cm) using hexane followed by hexane/EtOAc (6: 4) to furnish compound 7 (12 mg) [4]. Compound 8 (17 mg) [9] and compound 9 (100 mg) [13]. Compounds 10 (30 mg), 11 (50 mg) and 12 (64 mg) [14] were obtained from fraction DF-VII (22 g) after purification over CC using silica gel (200 g, column (50 × 4 cm) eluting with EtOAc/MeOH (9: 1). The structures of these compounds were established by their spectroscopic data and subsequent comparison with literature.
The pyridine solution of the compound (6) were treated with appropriate amount of acetic anhydride (as indicated in materials and methods section) and subjected to microwave treatment for sufficient time in 30 second intervals. After every 30 seconds a small portion of the reaction mixture was withdrawn and was analysed by TLC. All Conversions were completed with 2 to 4 doses of irradiation. Conventional acetylation was done with the same type of reaction mixtures by heating on a water-bath for one hour followed by keeping overnight. TLC indicated the presence of small amounts of unreacted compound in these reactions. Microwave assisted diacetylation of hennadiol afforded the corresponding diacetyl derivative (13) (56 mg) (Figures 1-4) while hydrogenolysis and subsequent separation of the resulting mixture by CC provided Lupeol (3) (17 mg) [4], compound (14) (10 mg) [15], and (15) (12 mg) [16] (Figures 2-4). All compounds obtained by chemical transformations were identified by examination of the 1H, 13C and 2D-NMR along with MS data.

Figure 1: Diacetylating reaction of hennadiol (6).

Figure 2: Structures of products obtained on hydrogenolysis of hennadiol (6).

Figure 3: Constituents from D. floribunda.

Figure 4: Constituents from chemical transformation of Hennadiol (6).
The crude extracts of different parts of the title plant were screened for antioxidant and lipoxygenase inhibitory activity while compounds 2-7, 10, 14 and 15 were subjected to antioxidant screening and urease inhibitory activity and the results are illustrated in Tables 1 and 2, respectively.
| S.No | Crude extract | Antioxidant Activity (% at conc 50 ug/10 ul) | Lipoxygenase Inhibition Activity IC50 (μM) |
|---|---|---|---|
| 1 | Blossom | 97.7 ± 0.70 at 50 ug | 68.2 ± 0.12 |
| 2 | Leaves | 96.6 ± 0.31 | 60.4 ± 0.76 |
| 3 | Stem barks | 57.7 ± 0.12 | 75.7 ± 0.54 |
| 4 | BHA | 44.2 ± 0.32 | - |
| 5 | Bacelin | - | 22.6 ± 0.17 |
BHA: Butylated hydroxyanisole (standard of antioxidant)
Bacelin: (standard of lipoxygenase)
Table 1: Results of antioxidant and lipoxygenase inhibition activity of crude extract.
| Compound | Antioxidant Activity IC50 (μM) | Urease Inhibition Activity IC50 (μM) |
|---|---|---|
| 2 | < 200 | 20.4 ± 0.76 |
| 3 | 65.3 ± 0.41 | Nil |
| 4 | 31.4 ± 0.33 | Nil |
| 5 | 43.1 ± 0.82 | Nil |
| 6 | 28.5 ± 0.08 | Nil |
| 7 | 16.7 ± 0.64 | 78.9 ± 0.75 |
| 10 | < 200 | 43.6 ± 0.23 |
| 14 | 54.1 ± 0.38 | Nil |
| 15 | 52.2± 0.18 | Nil |
| BHA | 44.2 ± 0.34 | - |
| Thiourea | - | 21.9 ± 0.32 |
BHA: Butylated hydroxyanisole (standard of antioxidant)
Thiourea: (standard inhibitor of urease)
Table 2: Results of antioxidant and urease inhibitory inhibition activity of pure isolates 2-7, 10, 14 and 15.
Authors are thankful to the International Canter for Chemical and Biological Sciences (ICCBS), University of Karachi, Pakistan for research facilities and Third World Academy of Sciences (TWAS), Italy for financial support under an ICCBS-TWAS Sandwich Program.
The authors declare no conflict of interest.
