Research Article - (2014) Volume 2, Issue 6
Ochna holtzii Gilg is a medicinal plant used extensively at the Kenya Coast for the treatment of various ailments. From solvent extracts of the root and stem barks of O. holtzii, seven constituents were isolated by standard chromatographic techniques (CC, VLC, prep-TLC and Sephadex LH-20). Their structures were analyzed by MS, UV,IR,1D and 2D NMR spectroscopy. All were found to be biflavonoids, including three novel compounds: dehydrate of lophirone C, hotzinol, and tri-O-methyl lophirone A. Crude methanol extracts of O. holtzii and the isolated biflavonoids were tested for antimicrobial activities against two Gram-positive bacteria, Staphylococcus aureus and Bacillus subtilis, two Gram-negative bacteria, Salmonella typhi and Pseudomonas aeruginosa, and the diploid fungus Candida albicans. The extracts and the isolated constituents showed varying levels of activities against the microbes. Afzelone D, lophirone A and the novel tri-O-methyl lophirone A showed strong activities against P. aeruginosa and S. aureus, with the latter being more susceptible. Calodenin B and the novel dehydrolophirone C also showed strong activities against these bacteria, but were more active against P. aeruginosa. However, none of these matched those of the antibiotic Chloramphenicol or the antifungal Fluconazole. This represents the first study on phytochemical and antimicrobial profiles of O. holtzii.
Keywords: Ochnaholtzii Gil; Ochnaceae; Biflavonoids; Dehydrolophirone C; Holtzinol, Afzelone D O-methylether
Ochna holtzii Gilg (Ochnaceae) is one of eight plant species of the genus Ochna that are found in Kenya [1]. Plants in this genus are known to be rich in biflavonoids, isoflavonoids and anthranoids [2-4]. Flavonoids exhibit a wide range of biological activities, and have been of particular interest as potential anticancer [5], anti-HIV-1 [6,7] and antibacterial agents and analgesics, as well as insecticides and insect antifeedants [8]. Moreover, flavonoids are effective antioxidants and antidotes for snake bites [9], and are considered to provide protection from cardiovascular diseases [10], certain forms of cancer and age related degeneration of cell components [11]. Ochnaholtzii is a plant found mainly at the coastal region of Kenya. The local communities use various parts of the plant (stem and root bark) to treat different health problems, including high fever, headaches and coughs, and to soothe persistent backaches, especially in old age [12]. It is also used extensively with other plant extracts in treating microbial infections [1]. The bio-activities of this plant and associated phytochemicals have not been systematically investigated. In the present study, we undertook chromatographic separation of solvent extracts of the stem and root bark, isolated and structurally characterized some of the constituents and compared their antimicrobial and antifungal activities with those of crude extracts. Here in we report our findings.
Compound 1 was obtained as yellow crystals from the EtOAc stem bark extract of O. holtzii. The UV spectrum exhibited absorption peaks at λmax 201, 223 and 274 nm, characteristic of compounds with highly conjugated system [13]. EI-MS (70eV) gave m/z at 526 [M+H]+ corresponding to C3OH20O8. This was further confirmed with NMR data including 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra. The 1H NMR spectrum displayed 15 signals of which thirteen (13) appeared in the aromatic region and 2 were olefinic with JH-H=15.4 Hz characteristic of a trans-configuration. The 13C NMR spectrum had 24 distinct signals for 30 carbon atoms, 2 of which were carbonyl carbon atoms and over 12 associated with several aromatic rings. Chemical shifts assignments were made from the analysis of 1H-1H COSY, HMBC and HMQC spectra. From the analysis of the 1H-1H COSY spectrum, the aromatic protons were distributed into one para-disubstituted, three ortho, para-trisubstituted aromatic ring systems and a dehydrobenzofuran moiety. The structure of compound 1 was confirmed from the analysis of its HMBC spectrum. The 1H and 13C NMR spectra of the compound closely compared with the spectral data of lophirone C [14], a compound isolated previously from Ochnaafzelii. The main difference was the presence of two quaternary carbon signals at δ156.2 and 114.5 at positions α2 and β2, respectively, in compound 1, indicating a complete furene ring, while in lophirone C, the two carbon signals were aliphatic at δ 88.6 and 57.6, respectively, as shown in (Table 1). Compound 1 was named dehydrolophirone C.
| Position | Compound 1 | Compound 3 | Compound 7 | |||
|---|---|---|---|---|---|---|
| δC | δH (ppm), m*, (J (Hz)) | δC | δH (ppm), m*, (J (Hz)) | δC | δH (ppm), m*, (J (Hz)) | |
| B1 – 1 | 114.7 s | 114.7 s | 117.0 s | |||
| B1 – 2 | 167.6 s | 167.7 s | 159.2 s | |||
| B1 – 3 | 103.8 d | 6.20 (1H, d, J=2.2 Hz) | 103.9 d | 6.27 (1H, d, J=2.2 Hz) | 103.3 d | 6.69 (1H, d, J=2.2 Hz) |
| B1 – 4 | 166.7 s | 166.7 s | 165.8 s | |||
| B1 – 5 | 109.3 d | 6.34 (1H, dd, J=9.0, 2.2 Hz) | 108.8 d | 6.42 (1H, dd, J=2.2, 9.1 Hz) | 116.1 d | 6.83 (1H, dd, J=2.2, 8.9 |
| B1 – 6 | 133.7 d | 7.91 (1H, d, J=9.0 Hz) | 133.2 d | 8.10 (1H, d, J=9.1 Hz) | 128.2 d | 7.85 (1H, d, J=8.9 Hz) |
| C1 | 193.3 s | 192. 8s | 177.1 s | |||
| α1 | 121.4 d | 7.70 (1H, d, J=15.4 Hz) | 119.3 d | 7.72 (1H, d, J=15.4 Hz) | 122.4 s | |
| β1 | 145.2 d | 7.83 (1H, d, J=15.4 Hz) | 144.8 d | 7.85 (1H, d, J=15.4 Hz) | 157.4 d | 8.22 (1H, s) |
| A1 – 1 | 130.8 s | 129.3 s | 135.7 s | |||
| A1 – 2 | 122.1 d | 7.71 (1H, d, J=2.0 Hz) | 126.9 s | 129.8 d | 7.16 (2H, d, J=8.4 Hz) | |
| A1 – 3 | 132.5 s | 132.1 s | 116.1 d | 6.56 (2H, d, J=8.4 Hz) | ||
| A1 – 4 | 158.2 s | 158.9 s | 157.4 s | |||
| A1 – 5 | 112.8 d | 7.58 (1H, d, J=8.8 Hz) | 111.0 d | 6.95 (1H, d, J=8.8 Hz) | 116.1 d | 6.56 (2H, d, J=8.4 Hz) |
| A1 – 6 | 126.8 d | 7.74 (1H, d, J=8.8, 2.0 Hz) | 132.1 d | 7.50 (1H, d, J=8.8 Hz) | 129.8 d | 7.16 (2H, d, J=8.4 Hz) |
| C2 | 196.1 s | 196.5 s | 204.8 s | |||
| α2 | 114.5 s | 113.3 s | 44.6 d | 6.02 (1H, d, J=12.2 Hz) | ||
| β2 | 156.2 s | 153.3 s | 54.3 d | 4.70 (1H, d, J=12.2 Hz) | ||
| A2 – 1 | 121.4 s | 135.8 s | ||||
| A2–2, 6 | 130.6 d | 7.48 (2H, d, J=8.4 Hz) | 130.5 d | 7.16 (2H, d, J=8.4 Hz) | ||
| A2-3, 5 | 116.7 d | 6.71 (2H, d, J=8.4 Hz) | 114.6 d | 6.56 (2H, d, J=8.4 Hz) | ||
| A2 – 4 | 160.8 s | 164.5 s | ||||
| B2 – 1 | 114.6 s | 115.3 s | 115.0 s | |||
| B2 – 2 | 167.5 s | 166.1 s | 166.8 s | |||
| B2 – 3 | 103.8 d | 6.27 (1H, d, J=2.2 Hz) | 103.3 d | 6.38 (1H, d, J=2.2 Hz) | 101.7 d | 6.28 (1H, d, J=2.3 Hz) |
| B2 – 4 | 167.3 s | 166.1 s | 168.0 s | |||
| B2 – 5 | 109.5 d | 6.05 (1H, dd, J=8.8, 2.2 Hz) | 109.0 d | 6.49 (1H, dd, J=2.2, 8.0 Hz) | 108.5 d | 6.34 (1H, dd, J=2.3, 9.0) |
| B2 – 6 | 136.7 d | 7.23 (1H, d, J=8.8 Hz) | 136.5 d | 7.97 (1H, d, J=8.0 Hz) | 134.4 d | 8.14 (1H, d, J=9.0 Hz) |
| A1–4 - OCH3 | 49.5 q | 3.76 (3H, s) | ||||
| A2-4 - OCH3 | 56.1 q | 3.76 (3H, s) | ||||
| B2-4 - OCH3 | 55.5 q | 3.66 (3H, s) | ||||
Table 1: 1H and 13C NMR data for compounds 1, 3 and 7.
Compound 2 was obtained as a yellow solid from stem bark. It gave m/z at 527 [M+H]+ corresponding to C3OH22O9 with EIMS (70eV), and MS fragmentation pattern, 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra identical to those of lophirone K [3]. Compound 3 was obtained from the stem bark as yellow crystals. It gave m/z at 449 [M + H]+ with EI-MS (70eV).The molecular formula was deduced as C24H16O9 and further confirmed by NMR data, including 1D NMR (1H,13Cs and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra. The 1H NMR spectrum displayed 10 signals and eight (8) of these appeared in the aromatic region.Eight of the ten signals (1D and 2D NMR) were very similar to those of α,β-unsaturated olefin moiety and B1 and B2 aromatic rings of lophirone C [14] and dehydrolophirone C. The main difference was absence in compound 3 of aromatic C-H signals corresponding to the para-disubstituted ring A2 (Table 1). In addition, lack of one aromatic proton associated with ring A1and presence of two doublets at δ 7.50 and 6.95 (J=8.8 Hz) showed the presence of a hydroxyl group at position 2 of the ring. These spectral features led to assignment of structure 3 for the compound which was named holtzinol.
Compound 4 was obtained as orange needles from the root bark that gave m/z 524 [M+H]+ corresponding to C30H20O9with EIMS (70eV), and MS fragmentation pattern, 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra identical to those of calodenin B [3].
Compound 5 was obtained from the root bark that gave m/z 511 [M+H]+ corresponding to C30H22O8with EIMS (70eV), and MS fragmentation pattern, 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra identical to those of lophirone A [15].
Compound 6 was obtained as a brown solid from stem bark that gave m/z 539 [M+H]+, corresponding toC32H26O8 with EIMS (70eV), MS fragmentation pattern, 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra identical to those ofafzelone D [14].
Compound 7 was obtained as yellow crystals from the EtOAc root bark extract. It gave m/z at 534 [M+H]+corresponding to C33H28O8, which was confirmed by NMR data, including 1D NMR (1H,13C and DEPT) and 2D NMR (COSY, HSQC, HMBC and NOESY) spectra. The 1H NMR spectrum of 7 displayed 19 signals. Two singlets at δ 3.76 and 3.66, integrated for three and six methoxyl protons, respectively. The six protons singlet indicated that the two methoxyl groups are symmetrically positioned. The other 17 signals were very similar with respect to shifts and splitting pattern with those of lophirone A [15] and afzelone D [16] reported earlier. The existence of three methoxyl groups implied that compound 7 was tri-methoxy derivative of lophirone A, that is, lophirone A trimethyl ether (Figure 1 and Table 1).
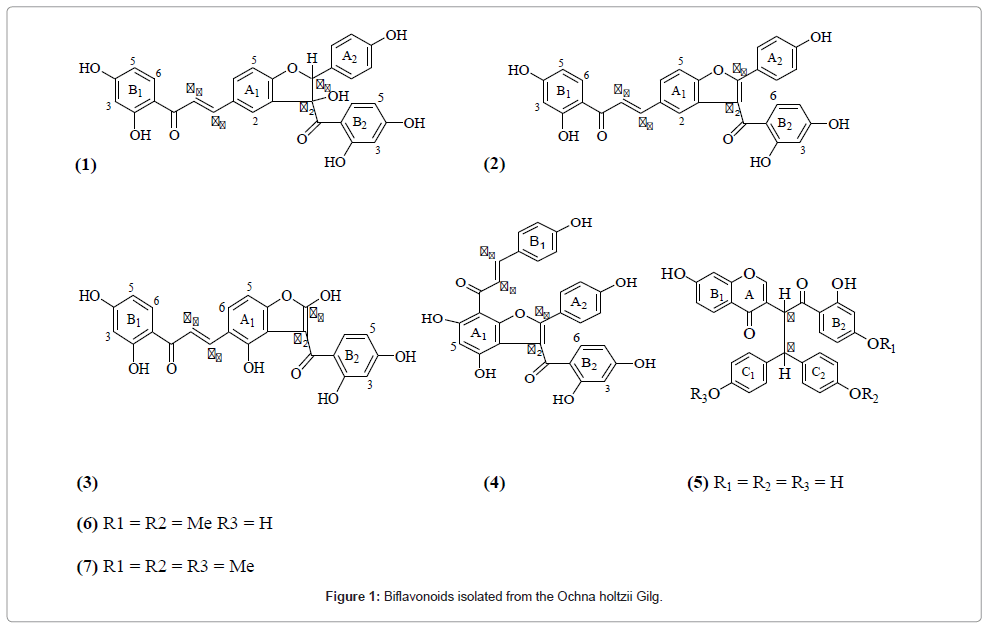
Figure 1: Biflavonoids isolated from the Ochna holtzii Gilg.
The root methanolic crude extract exhibited higher antimicrobial properties against S. aureus, P. aeruginosa and B. subtiliscompared to those of the stem bark extract (Table 2). The isolated compounds showed varying levels of activities against the two gram positive and two gram negative bacteria, as well as against the diploid fungus. Lophirone A, afzelone D and lophirone Atrimethyl ether also showed strong activities against these bacteria. Likewise, calodenin B and dehydrolophirone C showed also strong activities against the bacteria as well as against C. albicans. Further studies on related compounds that may be isolated from other Ochna and related plant species are expected to shed some light on the structural requirements for antimicrobial activities of this group of natural products. In addition, it would be interesting to extend the study to other bioactivities of these bioflavonoids. For example, calodenin B was previously found to exhibit cytotoxicity against breast cancer cells [17], and Lophirone A was reported to inhibit Epstein-Barr virus [18]. This biflavanoid has also been reported to exhibit potent cytotoxicity activity against melanoma (UACC62), renal (TK10) and breast (MCF10) cancer cell lines [19]. Recently, Ajiboye found that lophirone C has relatively high anticancer, antimutagenic, and antioxidant activities [20]. Thus, although our results with O. holtzii and its constituents provide some scientific rationale for the use of this ethno-medicinal plant by the communities of the coastal region of Kenya, it also suggests the need for extending the study to other Ochna species and other bioactivities.
| Isolate/extract | Micro-organisms | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S. aureus | P. aeruginosa | B. subtilis | S. typhi | C. albicans | ||||||
| (Z-I) | MIC | (Z-I) | MIC | (Z-I) | MIC | (Z-I) | MIC | (Z-I) | MIC | |
| Stem MeOH crude | 20 | 500 | 19 | 1000 | 23 | 250 | 17 | 1500 | 13 | NT |
| Root MeOHcrude | 26 | 250 | 22 | 500 | 26 | 200 | 16 | 1500 | 14 | NT |
| Dehydrolophirone C (1) | 21 | 250 | 24 | 500 | 23 | 250 | 15 | 1500 | 18 | NT |
| Holtzinol (3) | 16 | 1000 | 14 | 1500 | 16 | 2000 | 12 | 1500 | 10 | NT |
| Calodenin B | 20 | 250 | 25 | 250 | 23 | 250 | 15 | 1500 | 22 | NT |
| Lophirone A | 24 | 200 | 21 | 500 | 25 | 250 | 15 | 1500 | 17 | NT |
| Afzelone D | 23 | 200 | 20 | 500 | 25 | 250 | 15 | 1500 | 17 | NT |
| Tri-O-methylLophirone A (7) | 23 | 200 | 20 | 500 | 24 | 250 | 14 | 1500 | 16 | NT |
| Chloramphenicol | 32 | 36 | 31 | 59 | 32 | 48 | 20 | 200 | - | - |
| Fluconazole | - | - | - | - | - | - | - | - | 26 | 72 |
Table 2: Anti-microbial activities of methanolic extracts of root and stem barks of O. holtzii and isolated compounds.
Spectral measurements
1H (1D, 2D COSY) and 13C spectra were recorded using Varian Gemini 400 MHz (NMR) instrument using CD3OD as solvent. Peaks on 1H-NMR were recorded as singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q) and (or) broad (b) using internal standard TMS as reference. The 13C-NMR multiplicity was determined by DEPT experiments. Chemical shifts were recorded in δ (ppm) and coupling constants, J, in Hertz (Hz). Standard sequences were used for COSY, HMQC, HMBC and NOESY experiments. IR: KBr pellets technique.
Chromatographic techniques
These include CC: silica gel 60 (0.063-0.200 mm, Merck); VLC and TLC: silica gel 60 F254 (Merck) precoated aluminum plates; Sephadex LH-20 used as filter gel; and p-anisaldehyde spray with UV-Vis was used for visualization.
Disc diffusion assay
The crude extracts and the isolated compounds were tested for antimicrobial activities using agar diffusion technique against two Gram-positive, Bacillus subtilis and Staphylococcus aureus, two Gramnegative, Salmonella typhi (Type K [1]) and Pseudomonas aeruginosa, and a yeast Candida albicans. The plate diffusion method was used. Chloramphenicol was used as positive control. 14 g of nutrient agar was dissolved in 0.5l of distilled water. Round filter paper MN 615 of 9 cm diameter was punched and the paper pieces sterilized. Nutrient agar (15 ml) was poured into Petri dishes in a lamina flow apparatus under sterile conditions. Then 0.1 ml of bacterial solution was added to it. Filter paper pieces containing 100 μg of the test extract were put on Petri dish and then finally incubated at 37°C for bacteria or 30°C for fungi. The results observed were recorded by measuring the diameter of the zone of inhibition from original 6 mm.
Determination of MIC
The MIC was determined using two-fold serial dilution method in a peptone water solution for bacterial and PDA broth for yeast and fungal of the active extracts. Each tube was then inoculated with 0.1 ml of standardized bacterial suspension (1×108CFU/ml) and fungal suspension (1×108 spores/ml). The cultures were then incubated at 37°C for 24 hours for bacteria, 48 hours for yeast and at 30°C for 72 hours for fungi. The first tube showing no growth was the MIC.
The stem and root barks of O. holtzii were collected from Arabuko- Sokoke Forest, Malindi, Kilifi County of Coast province in Kenya in April 2010. The plant was authenticated by Mr. Lucas Karimi, Department of Complementary Medicine, Kenyatta University. A voucher specimen (No. MM/002/10) was deposited at the herbarium of the Kenya National Museums, Nairobi.
Method of extraction and isolation
Air-dried stem and root barks of O. holtzii were separately ground to give fine powders (5.58 and 7.23 kg, respectively) and each was extracted separately using solvents of increasing polarity sequentially (starting with hexane, then DCM, followed by EtOAc and finally MeOH) for 48 hrs each with occasional swirling to ensure thorough extraction. The extracts were decanted and filtered through Whattman filter paper and the marcerate steeped in solvent again for 48 hrs. The extraction process was repeated 3 times when a clear extract was obtained. The filtrates were combined and concentrated using rotary evaporator under reduced pressure and a temperature of 45°C. A small portion of each crude extract was used for bioassays. Evaporation of the solvent from the stem bark ethyl acetate extract yielded 40 g of a dark brown residue. This extract was subjected to fractionation by Vacuum Liquid Chromatography (VLC) on silica gel with a Hexane:DCM - DCM:MeOH gradient (100:0-0:100) to yield 172 × 50 ml fractions. Fractions were combined after TLC comparison, which were then fractionated by column chromatography on silica gel using a gradient mixture of DCM/MeOH, starting from pure DCM, followed by 50:50 blends to pure MeOH. Fractions of 50 ml each were collected and these were subjected to further purification using Sephadex (L-20) column 1:1 (DCM:MeOH)and prep-TLC with the combined fractions using 1:1 (DCM:MeOH). Through this procedure, compound 1 was obtained. Using same procedure, other compounds isolated from the stem were lophirone K (2) [3] and afzelone D (6) [16]. A similar procedure was used with the 9 g root bark ethyl acetate extract and fractions were combined on the basis of TLC comparison and subjected to further purification. Thus, calodenin B (4)[3], lophirone A (5)[13], and compounds 3 and 7 were isolated from the root extract.
Dehydrolophirone C (1)
Yellow crystals, MP 198-200°C
IR (KBr): 2926 (br), 3356, 1620, 1512, 2962 cm–1.
UV λmax 201, 223 and 274 nm
1H and 13C NMR: Table 1.
EI-MS m/z (%) 207 (100), 509 (1, [M+H]+), 433 (2), 417 (3), 389 (4), 355 (5), 281 (24), 259 (5), 241 (8), 222 (5), 191 (10), 147 (21), 133 (7), 129 (100), 112 (19), 96 (8), 83 (12), 73 (9), , 55 (2).
Holtzinol (3)
Yellow crystals, MP 178-180°C
UV λmax : 201, 224 and 274 nm
IR (KBr): 2926 (br), 3410, 2962, 1627, 1512 cm–1.
1H and 13C NMR: Table 1.
EI-MS m/z (%) 207 (100), 449 (1, [M+H]+), 429 (2), 390 (3), 355 (5), 281 (24), 222 (5), 259 (5), 241 (8), 191 (15), 147 (21), 133 (7), 129 (100), 112 (19), 96 (8), 83 (12), 73 (9), 71 (22), 57 (30), 55 (2).
Lophirone A trimethyl ether (7)
Yellow crystals.MP 192-194°C.
UV λmax : 218, 282 nm
IR (KBr): 2926 (br), 3256, 2963, 1605, 1512 cm–1.
1H and 13C NMR: Table 1.
EI-MS m/z (%) 207 (100), 534 (2, [M+H]+), 428 (1), 355 (4), 341 (2), 306 (19), 281 (26), 214 (100), 197 (12), 183 (41), 181 (18), 163 (3), 152 (11), 149 (6), 135 (9), 133 (10), 121 (23), 108 (30), 94 (65), 77 (10), 73 (9), 65 (17), 55 (7).
We are grateful to Kenyatta University and its technical staff for providing space and some apparatus used in this work. We thank AICAD (the African Institute for Capacity Development) for partial funding of the project. We also appreciate Mr. Lucas Karimi, Department of Pharmacy and Complementary Medicine for authenticating the plant.
