Research Article - (2015) Volume 3, Issue 3
Camptothecin is one of the human success stories in the history of drug discovery from natural resources. In this mini-review, the painstaking efforts of eminent scientists of different disciplines to isolate and identify camptothecin as active anticancer compound from Camptotheca acuminata are narrated. Due course, the unique mode of action of camptothecin was discovered. Clinically some tumors acquire de novo resistance to camptothecin and a few of molecular mechanisms responsible for clinical resistance are discussed in brief. Finally, we discuss novel sources and how biotechnological methods are utilized for a sustainable production of camptothecin without impacting the natural flora.
Plants are always considered as a treasure trove of medicinal compounds to alleviate the human ailments [1]. Even the advent of industrialization and discovery of synthetic pharmacological chemicals, the interest on natural products as a source for potential therapeutic chemical entities has not been diminished. Nature still serves as the only source for many complex natural chemical compounds because the inability to synthetically produce them in the laboratory. Many natural products form the basic lead molecules for the synthesis of derivatives which are of pharmaceutical significance. Therefore, the search for novel compounds against various illnesses is always a hot topic of biomedical research [2-5]. During such a drug discovery research program set up by National Cancer Institute (NCI), two natural products were identified as potent anticancer compounds, named camptothecin and taxol [6,7]. These compounds, because of their unique mode of action, became promising lead molecules for treating different forms of malignancies in later years. In this mini review, a short history of isolation of camptothecin, its mode of action at molecular level, mechanisms of resistance to camptothecin and alternative sources of its isolation are discussed.
Camptothecin
Camptothecin, (C20H10N2O4; molecular weight: 348.352 g/ mol; IUPAC name: (S)-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7] indolizino[1,2-b]quinoline-3,14-(4H,12H)-dione) is a cytotoxic monoterpenoid indole alkaloid and structurally a planar heteropentacyclic ring, that includes a pyrrolo [3,4-beta]-quinoline moiety (rings A, B and C), conjugated pyridone moiety (ring D) and one chiral center at position 20 within the alpha-hydroxy lactone ring with (S) configuration (the E-ring) (Figure 1). The planar structure is believed to contribute to the inhibition of topoisomerase function [8].
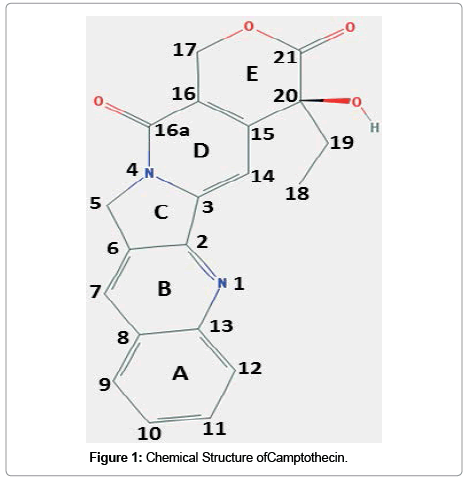
Figure 1: Chemical Structure ofCamptothecin.
A historical account on discovery of camptothecin
In 1955, National Cancer Institute (NCI) created a Cancer Chemotherapy National Service Center (CCNSC) for the evaluation for anticancer efficacy of potential compounds submitted by drug companies and institutions. Initially, CCNSC was screening chemically known structures and by 1960 they started to screen natural products of unknown chemical ingredients from plant and animal origin. The NCI plant program was led by Jonathan Hartwell, an eminent organic chemist, and he later compiled the documents of traditional use of plants to treat cancer from ancient forms of medical practices from Egypt, China, Greece and Rome [9-12].
The United States Department of Agriculture (USDA) was providing plants to NCI for anticancer screening. At Wisconsin Alumni Research Foundation, the plants were extracted and sent to different laboratories to evaluate their potential to kill cancer cells. The samples were initially screened for their cytotoxic effects in KB (oral epidermoid carcinoma) cell culture, a cell line from a human cancer. They were also tested in three tumor xenograft mouse models for S-180, a sarcoma; CA-755, an adenocarcinoma; and L1210, a lymphoid leukemia. Those crude extracts showed anticancer properties were then fractionated for isolating the active compounds at three different laboratories. One among them was newly established Research Triangle Institute (RTI), North Carolina. Manroe E. Wall was leading the Natural Product Laboratory in RTI. Of thousand ethanolic plant extracts screened by NCI for anticancer efficacy, extract of Camptotheca acuminata, a tree native to China, showed very potent cytotoxic activity in L1210 mouse leukemia cell assay, to which not any other plant extracts were active. Different fractions of crude extracts of C. acuminata were studied for life prolongation in mice harboring L1210 leukemia, and KB in vitro assay. Wall was ambitious to identify the active compound responsible for anticancer activity in the extract of C. acuminata. By this activity based fractionation strategy, they finally isolated the pure compound responsible for killing L1210 leukemic cells, and they termed this compound as camptothecin in 1966. This newly identified indole alkaloid was not only active against L1210, but also found to be cytotoxic against p338 leukemic cells [6]. Keith Palmer and Harold Taylor did the original isolation of camptothecin at RTI, and Ed Cook worked on determining its structure. By the instruction of Wall, Wani made a camptothecin derivative and sent to Andrew McPhail and George Sim at the University of Illinois who reported the tentative structure of camptothecin. This remarkable achievement was published in 1966 in the Journal of the American Chemical Society, the first paper Wall, Wani and colleagues published on a natural product with anticancer potential [6].
Identification of mode of action of camptothecin
Preliminary works of Moore BG [13], Kessel D [14,15], Bosmann HB [16], Horwitz, SB [17,18] and others have established that the synthesis of macromolecules such as RNA, DNA and protein are inhibited by camptothecin in tumor or dividing mammalian cells. The inhibition over DNA synthesis was transient when the drug is removed. But prolonged incubation could lead to irreversible suppression of DNA synthesis. Also camptothecin was not able to bind with isolated DNA when incubate them together. But, when intact cells were incubated with camptothecin for a short term, it induced rapid but reversible fragmentation of cellular DNA. The longer the incubation, more the damage of DNA that leads to cell death. However camptothecin was found to be ineffective to block the effects of purified RNA and DNA polymerases. Later studies by Hsiang confirmed topoisomerase 1, not topoisomerase 2, is the molecular target of camptothecin [19-21].
Topoisomerases (topo) are enzymes that relieve the torsional strain associated with the unwinding of DNA while replication or transcription [22]. There are two main forms of topoisomerases called topoisomerase 1 and topoisomerase 2 based on whether they cut single or both strands of DNA. During DNA replication or transcription in cells, topo1 forms covalent complex with DNA and this complex act as a target for camptothecin or its derivatives to form a topo1-camptothecin-DNA ternary complex [22,23]. It is suggested that camptothecin inhibiting the religation step of cleavage/religation reaction during the process of replication or transcription. X-ray crystallographic structure of DNAtopo1 complexes is well-studied [24,25], and crystallization of a topo1- camptothecin-DNA ternary complex has provided some light into the molecular interaction among these three units in the ternary complex [22]. The mechanism of camptothecin-induced cell death is dependent on synthetic phase (S-phase) of cell cycle [22,26]. A replication fork collision model has been proposed to explain S-phase dependent cytotoxicity induced by camptothecin, since the reversible ternary complex cannot induce cell death itself. But, when the separation of DNA advances and replication fork collide with topo1-camptothecin- DNA ternary complex, strand break is induced and drives cell death [22]. There are three biochemical events have been noted after replication fork collision, (a) the formation of double strand breaks, (b) irreversible arrest of replication fork and (c) the formation of top1- linked DNA break at the site of collision. The relative contributions of each of these events to cell death are not well-understood yet [27-29].
Interfacial inhibitor concept
Certain small molecular natural products can interfere a biological function that otherwise progress without inhibition as the result of macromolecular assemblage [30]. For example, DNA replication is a complex biological process required the concerted and fast moving action of different macromolecules including DNA, topoisomerases, and polymerases. Interfacial inhibitors intrude to bind with high selectivity to a binding site within macromolecular complexes. As in the case of camptothecin, it intrudes into the macromolecular assembly of DNA and topoisomerase 1 that essentially stalls the kinetics of untwisting of DNA during replication. This type of inhibition introduced by small molecules sneaked into the interface between two or more macromolecules is referred as interfacial inhibition [31]. The interaction of camptothecin with Topo1 enzyme is established by Hydrogen bonding that is depicted in (Figure 2). Camptothecin establish bonding with DNA by π-π electron interactions between the heterocyclic ring system of the drug and the nucleotide bases flanking the cleavage site which hinders the rotation of the DNA [32,33]. This interference inhibit strand passage or torsion release. When replication fork advances and collide with Topo I-camptothecin-DNA complex, result in double stand breaks and DNA damage which ultimately leads to cell death. Besides camptothecin, several other natural products including forskolin, tubulin inhibitors and immunophilins target protein interfaces [31].
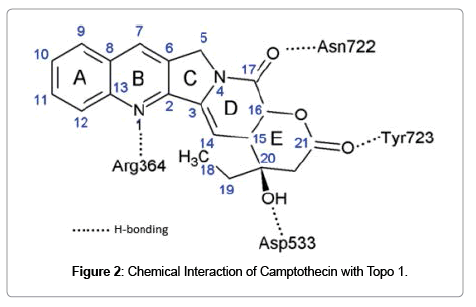
Figure 2: Chemical Interaction of Camptothecin with Topo 1.
Clinically useful derivatives of camptothecin
Although identified in 1958 and soon commenced anticancer clinical trials, further endeavors with camptothecin were discontinued pertained to its poor water solubility along with off-target toxicities such as myelosuppression, diarrhea and hemorrhagic cystitis [34]. The interest in this compound was rekindled after Topo1 was found the molecular target for camptothecin. Chemical derivatives created by modifying rings of camptothecin improved the pharmacokinetics and toxicity profiles of the parent compound. Currently two derivatives of camptothecin approved by food and drug administration (FDA) for clinical practice are topotecan and irinotecan. Topotecan is commercially known as Hycamtin® that used as second-line chemotherapeutic drug for cervical, ovarian, and small cell lung cancer. Irinotecan is sold in the brand name of Camptosar® for the treatment of metastatic colorectal cancer. Besides these FDA-approved camptothecin derivatives, there is a line of candidate molecules of same class under investigational category.
Topotecan
It is a water soluble derivative of camptothecin with substitutions in ring A; an alkylamine at position 9 and a hydroxyl at position 10 (Figure 3a). Followed the promising results obtained from phase I trial with topotecan [35], a dose of 1.5 mg/m2/day was selected for phase II/III evaluation. This regimen has been shown to have activity in recurrent ovarian cancer [36], relapsed small cell lung cancer [37], non-small cell lung cancer [38,39], and breast cancer [40]. Topotecan has been found effective in combinatorial treatment with pegylated liposomal doxorubicin in brain tumor [41], platinum resistant ovarian and peritoneal cancer [42,43].
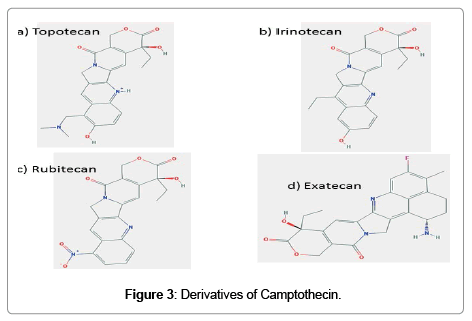
Figure 3: Derivatives of Camptothecin.
Irinotecan
During development, it was known as CPT-11. It has a bipiperidine carboxylic acid side chain at position 10 of ring A and an ethylene group at position 7 of rings B (Figure 3b). The active form of irinotecan is a hydrolytic product, SN-38. This is then inactivated by glucuronidation by uridine diphosphate glucoronosyltransferase 1A1 (UGT1A1). Irinotecan is a very active chemotherapeutic agent used for the treatment of several malignancies, including colorectal cancer, gastroesophageal tumors, lung cancer, breast cancer, ovarian cancer, and primary brain tumors. It used as a component in combinatorial regimen FOLFIRI which contains infusional 5-fluorouracil and leucovorin also.
Etirinotecan pegol, a conjugate of Irinotecan
The 7-ethyl 10-hydroxy camptothecin is the active metabolite of irinotecan and this metabolite causes the treatment-associated diarrhea and neutropenia. In order to overcome this drug related adverse effects, certain modifications are being utilized. In pegylation, the active compound is linked to one or more molecules of polyethylene glycol (PEG) which covers the active compound and imparts more solubility, retention and bioavailability [44].
In phase I study, etirinotecan pegol for 3 dosing schedules (58-245 mg/m2) at 3 times every 4 weeks, or 1 time every 14 days, or 21 days were infused into patients with refractory malignant solid tumors. Substantial anticancer activity was observed despite manageable gastrointestinal disorders. The maximum tolerable dose suggested for phase II study was 145 mg/m2 every 14 days and 21 days [45].
In a phase II, patients with platinum resistant refractory ovarian carcinoma were received 145 mg/m2 etirinotecan pegol every 14 days or 21 days. The treatment continued until clinical goal achieved or adverse events are reported. Both schedules provided significant objective response rate and progression free survival, being once in 21 day schedule was slightly superior to 14 day schedule [46].
Analogues of camptothecin under clinical investigation
Rubitecan
In rubitecan, hydrogen is replaced by a nitro group at position 9 of the ring A of camptothecin (Figure 3c). Preclinical studies using human cancer xenografts in nude mice with maximum tolerable dose of rubitecan has shown 100% growth inhibition of 30/30 tumors tested and in 24/30 in their total disappearance. These 30 tumors comprised all the most common human cancers: lung, colorectal, breast, pancreatic, ovarian, prostate, stomach, melanoma and leukemia [47]. Despite this promising preclinical success, treatment with rubitecan has exhibited disappointing results in small phase I/II trials against a number of solid tumors [48-50] with the exception of pancreatic cancer [51]. Results of clinical trials in the next few years should determine whether rubitecan can find a role in cancer therapy.
Exatecan
It is a completely synthetic analogue of camptothecin (Figure 3d). This compound has increased aqueous solubility, greater tumor efficacy, and toxicity is manageable. In preclinical studies, exatecan was found active against multiple cancer cell lines and human breast, gastric, renal colon, ovarian, cervical and lung xenografts models in mice. As p-glycoprotein multidrug transporter pump does not recognize exatecan, contribute to its activity, at least in part. It is equally effective in both human lung cancer PC-6 and the cell variant that over expresses P-glycoprotein. However, phase I/II studies have not provided significant activity in many forms of human cancers [52-56]. But in some clinical trials, as in previously untreated metastatic gastric cancer and biliary tract cancer, modest or minimal improvement was noted [57,58]. No clinical studies have published with exatecan since 2007.
FL118, a novel structurally similar analogue of camptothecin
Although FL118 (Figure 4) shows structural similarity to topotecan and irinotecan, this was not synthesized using camptothecin as template. It was found out through a high-throughput screening of compound libraries for inhibition of survivin, an antiapoptotic protein [59]. The effectiveness of FL118 at nanomolar concentration, compared to SN-38 at 1μM concentration, to inhibit topoisomerase 1 is p53 independent. But in colorectal cancer cells expressing wild type p53, FL118 activates p53 signaling by specifically inhibiting p53 polyubiquitination and monoubiquitination by Mdm2-MdmX E3 complex formation, which leads to senescence. But even if MdmX is overexpressed in these cells, FL118 can alternatively induce apoptosis in a p53-independent mechanism [60]. In addition, FL118 not only inhibit antiapoptotic proteins like survivin, XIAP, cIAP2 and Mcl-1 while inducing the expression of pro-apoptotic proteins Bax and Bim. FL118 exhibits superior antitumor efficacy in mouse bearing human tumor xenograft compared to irinotecan, topotecan, doxorubicin, 5-FU, gemcitabine, docetaxel, oxaliplatin, cytoxan and cisplatin. One of the reasons for the resistance to irinotecan and topotecan is the overexpression of efflux pump ABCG2. Interestingly, FL118 is not an ABCG2 substrate, which fails the cancer cells to efflux FL118. Recently, intravenous administration of a Tween-80 free formulation of FL118 was found to be much more effective in eliminating human tumor xenografts in mice which make it a safer candidate for clinical trials. Taken into consideration, the molecular targets and superior antitumor activity place FL118 a promising candidate for clinical trials [61].
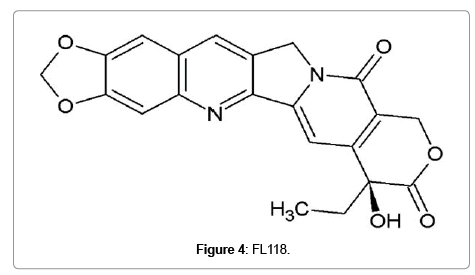
Figure 4: FL118.
Because of the unique mode of action of topo1 poisoning, a broadspectrum of cancers is being treated by camptothecin derivatives such as topotecan, irinotecan, 9-aminocamptothecin or exatecan. Despite the fact that it is widely used anticancer drug, the acquired clinical resistance towards treatment is the challenge faced by health care professionals. This drug resistance can have it origin from either pharmacological modification of drug in tumor environment or entirely due to overexpression of proteins related to tumor cell survival [62-68]. Yeast and mammalian cell culture models suggest the mechanism of resistance may be related to lesser accumulation of drug, because of changes in the expression of proteins of cellular drug uptake, or metabolism, or efflux. The other mechanisms may be alteration in topoisomerase I, or a different response to cleavable topo1- camptothecin-DNA complex. Here we review the above mentioned mechanisms briefly [63].
The efficacy of drug is dependent upon the balance between active cellular uptake, metabolism and efflux mechanisms. In case of oral formulations of camptothecin, both active and passive transport mechanisms are involved in intestinal absorption. The overexpression or activity of p-glycoprotein (P-gp), or multidrug-resistance-associated protein (MRP) in the intestinal epithelia can limit the oral absorption of camptothecin-11 [69]. The active form of camptothecin-11 is SN-38 which in turn formed by the action of carboxylesterase [70]. The alteration of this enzyme can decrease the sensitivity of camptothecin to cancer cells. Furthermore, glucuronidation of SN-38 by liver glucuronidases can increase the efflux which is associated with resistance to colon, breast and lung cancer cells [71-73]. Another efflux protein which involved in camptothecin resistance is BCRP [72], but some derivatives of camptothecin, for example lipophilic 7-modified camptothecin, are not a substrate for this protein [74].
The second mechanism proposed for camptothecin resistance was mutations in topo1 [75]. Different point mutations, especially near the site of catalytic tyrosine position at 723, and sites which bind with DNA and are in close vicinity of intercalated camptothecin are also implicated in drug resistance [22]. Interactions of topo1 and other proteins like nucleolin can affect camptothecin sensitivity [76]. The function of nucleolin is to bind to topo-1 and recruit to nucleolus for transcription. The absence of nucleolin can diffusely scatter topo1 in nucleus than in a localized distribution in nucleolus [77,78]. Also in camptothecin resistant cells, SUMO (small ubiquitin-like modifier) can modify topo1 and reduce the interaction of topo1-DNA, thereby minimizing the topo1 mediated DNA damage by camptothecin [76].
The third mechanism is the alteration in the cellular response to topo1-camptothecin-DNA ternary complex. A variety of proteins involved in DNA replication, cell cycle checkpoints, or DNA repair are implicated in camptothecin sensitivity. Examples are Chk1, ATM, ATR, RAD9, Dpb11p, MSH2 [26,79-82]; and their absence or loss of function can increase the camptothecin sensitivity. Also pro-apoptotic proteins like p53, Bax and anti-apoptotic Bcl-2 levels were altered in camptothecin resistant cancers [83-85].
The original source of camptothecin was C. acuminata and the increased demand of camptothecin as a lead molecule for the production of anticancer-derivatives persuaded researchers to explore novel sources for a sustainable production. Young leaves are the largest source of camptothecin (4-5 mg/g dry weight) from C. acuminata compared to other parts [86]. Later Govindachari and Viswanathan reported that Nothopodytes foetida (Mapia foetida) is a promotable source of camptothecin in large quantities [87]. Now the presence of camptothecin has been recognized in many non-related angiosperms as well as an endophytic fungus, LY357 [88]. Various species of Ophiorrhiza, such as O. pumila, O. rugosa, and O. liukiuensis have been identified as natural sources of camptothecin [89-92].
The dependence on natural flora for camptothecin in fact threatens their existence and it becomes inevitable to find sustainable alternatives. In vitro plant cell and tissue cultures have been utilized as a sustainable source of drug leads. Biotechnological methods offer development of medicinal plants in vitro as well as bioreactors for the production of natural products for the preparation of pharmaceuticals. The initial attempt to produce camptothecin from cell suspension cultures of C. acuminata was reported by Sakata in 1974 [93]. However a satisfactory level of production of camptothecin was not achieved by this method. But the shoot cultures of C. acuminata compared to cell suspension found to produce higher amounts of camptothecin [94]. Later, in this aspect Roja and Heble established in vitro cultures of N. foetida and confirmed the presence of camptothecin by HPLC methods [95]. Recently, plant biotechnologists have turned their interest to establishing shoot and root cultures of different species of Ophiorrhiza for a continuous source of camptothecin.
Shoot and root cultures of Ophiorrhiza species for camptothecin
The identification that different species of Ophiorrhiza produce camptothecin, propelled endeavors to establish tissue cultures and micro-propagation of these herbaceous plants for an alternate source of camptothecin. In 2001, Saito et al., established hairy root cultures of O. pumila transformed by Agrobacterium rhizogens strain 15834. Transformation increased growth rate of roots and treatment of these roots with elicitors, such as yeast extract, salicylic acid or methyl jasmonate, and precursor feeding with tryptophan or secologanin positively affected the camptothecin production [96]. Cloning and characterization of cDNAs encoding strictosidine synthase (STR) and tryptophan decarboxylase (TDC), two key enzymes in the biosynthesis of terpenoid indole alkaloid, in hairy roots provided evidence for their role in camptothecin biosynthesis. The high expression of STR and TDC cDNA observed in hairy roots, roots and stems were closely correlated with STR protein accumulation as observed by immunoblot analysis. Plant stress compounds like salicylic acid repressed expression of STR and TDC, suggesting coordinate regulation of these genes for camptothecin biosynthesis [97].
There are two other species of Ophiorrhiza, O. rugosa and O. alata were transfected to produce hairy roots. In O. rugosa, A. rhizogens strain LBA9402 was used to transfect. These roots when exposed to light, spontaneous regeneration of shoots were observed. The quantitation of camptothecin revealed that hairy roots contain 0.009% of dry weight; and shoots contain 0.012% of dry weight. In O. alata, A. rhizogens TISTR 1450 was used to transfect nodal explants in half strength Murashige and Skoog medium. HPLC analysis revealed camptothecin content was 0.079% of dry weight in these hairy roots of O. alata.
Recently, a metabolic engineering strategy was adopted to enhance the camptothecin content in hairy roots of O. pumila. STR and geraniol 10-hydroxylase (GH) genes were isolated from Catharanthus roseus introduced into hairy roots of O. pumila, either singly or together. When the genes are over-expressed individually, the GH increased camptothecin content better than STR. In contrast, simultaneous expression of both genes synergistically increased camptothecin production. This opens new avenues of creation of transgenic plants those produce superior amounts of camptothecin without violating natural flora [98].
Camptothecin was originally discovered as an anticancer compound. Soon it was found that, besides the anticancer activity, camptothecin derivatives exhibit potent anti-HIV efficacy also [99]. Many investigational derivatives of camptothecin are now in clinical trials including exatecan and rubitecan, which will potentially increase demand for these drugs in the future. In 2008 alone, the annual trade of irinotecan and topotecan together had reached 2.2 billion US dollars [100]. The annual production of camptothecin throughout the world is only 600 kg, which cannot meet the demand estimated to be approximately 3000 kg/year in the international market. Although, tremendous research efforts are being made to enhance the in vitro production of camptothecin, unfortunately none of the methods has come out of the laboratory for a commercial scale production. This underscores more efforts are required for the identification of economically feasible methods which can successfully substitute or append to the current camptothecin production line.
