Research Article - (2015) Volume 3, Issue 6
Latex of F. botryocarpa fruit is applied on skin infections in Papua New Guinea ethnotherapeutic practices. Systematic bioassay guided separation and isolation of subsequent fractions of latex extracts resulted in three bioactive fractions. Structures were determined by physical (M.pt and Rf values) and spectroscopic (1D-1H NMR, 2D-HSQC NMR, 2D-HMBC NMR) and MS ESI-POS. The two methylene protons (2H-1) and 2H-3) resonate as triplets at δ 3.59 and δ 4.99 respectively. Electron dense δ 4.99 (2H-3) on (C-3) depicts the strong electron withdrawing component, quaternary nitrogen (=N=+). Protons resonating at δ 3.88 and δ3.89 are singlets depicting two methoxy groups. Both δ 3.88 and δ 3.89 are para-aryls substituents. All isolates, (1), (2) and (3) were identified to be ficuseptine. 2D-NMR and MS of (2) found it to be ficuseptine chloride “2, 3-dihydro-6, 8-bis (4-methoxyphenyl)-1,2,3-trihydroindolizidinium chloride”. The counter ions were not established and provide promising lead for further investigation.
Keywords: Ficus botryocarpa; Latex; Antimicrobial activity; Ficuseptine; Skin infection
Fruit latex of F. botryocarpa was used to treat sores, wounds and other superficial skin infections in East Sepik Province, Papua New Guinea (PNG) [1-3]. Staphylococci are the principal causative agents accounting for 30% to 50% of wound infections [4]. Preliminary antimicrobial screening has shown activity of methanol extract of latex (FL) on Staphylococcus albus and Staphylococcus aureus with inhibition zones of 28 mm and 26 mm respectively [5-7].
Investigation of the active constituents from the latex of F. botryocarpa centered on alkaloids. Raw fruit latex was extracted with methanol and filtered separating the bioactive filtrate (F2) from the non-bioactive filtered (F1). (F2) was confirmed as predominantly alkaloids by Dragendorff and Mayers test [8]. By means of preparative chromatography three fractions, (1), (2) and (3) were isolated from (F2). These fractions showed strong activity against Escherichia coli, S. aureus and two bacterial colonies types isolated from an infected folliculitis [9]. This study attempts to identify chemical structures of these alkaloid isolates.
Sampling
Pricked fruits with latex protruding from petiole and fruit bud were collected in 40 ml polyethylene sampling bottles at Bulolo, PNG.
Sample preparation
Combined latex, 18 ml was extracted with 50 ml methanol and subject to vibration in a flask shaker (Stuart FS-1) at 200 oscillations per minute for 20 minutes. The process was repeated three times to obtain the primary extract (FL). Insoluble (F1) and soluble (F2) fractions were recovered by gravity filtration using 125 mm and 185 mm using whatman filter papers and concentrated on a BUCHI rotary evaporator under reduced pressure followed by drying under vacuum.
White gummy-like substance, were collected as filtered (F1) and air dried at room condition to solidify. The soluble filtrate (F2) was concentrated on a BUCHI rotary evaporator under reduced pressure followed by vacuum drying. (F2) gave a golden - yellow paste.
1 g of F2 was dissolved in 10 ml MeOH and deposited on a horizontal thin line at the bottom of a preparative silica gel plate. The plate was placed in a chamber containing the solvent, acetone/toluene/ ethanol/ammonia (40:40:6:2) mixture [10]. Three spots of Rf0.26, Rf0.14 and Rf0.07 resembling bands developed earlier were ideally developed (blue fluorescence) and marked under UV light at 254 nm. The three fractions were scraped off the glass plate (20 × 20 cm) into separate screw cap conical flask (100 ml). Each fraction was dissolved in Me OH (3 × 30 ml) vibrated at 200 oscillations per minute for 10 minutes and filtered. The filtrates were concentrated in vacuo at 68°C (BUCHI) and further vacuum dried yielding fraction (1) 2.10 mg, (2) 4.20 mg and (3) 6.13 mg.
Instruments
Melting Point Instrument, Digital 9000 Series, IA9100 230 V (max. 370°C), Clarkson, USA. Melting points were determined by inserting glass capillaries filled with samples to electrothermal melting point apparatus hovels and the values are uncorrected.
NMR spectra were obtained using Varian Unity Inova 300 MHz using VNMRJ software and Bruker Avance spectrometer (600 MHz) equipped with a cryoprobe operated using Top Spin software. CD3OD used as reference (1D, δ=49.0 ppm (13C) and 3.31 ppm (1H), whilst 2D δ=0.00 (1H) ppm).
High resolution mass spectral data were determined on a Bruker BioApex 47 Fourier transform mass spectrometer (FTMS) with an electrospray (ESI) all event sequences were controlled and data reduction performed using Bruker Daltonics XMASS version 7.0.3.0 software.
The three fractions showed distinct separation on the chromatogram evident with M.pt and Rf values (Table 1). However, 1H-NMR spectral information of the three isolates corresponded with literature information of ficuseptine chloride, 2, 3-dihydro-6, 8-bis (4-methoxyphenyl)-, 1H-Indolizinium chloride, (Figure 1).
| Fraction | M.pt (°C) | Rf |
|---|---|---|
| 1 | 220-221 | 0.26 |
| 2 | 213-215 | 0.14 |
| 3 | 218-220 | 0.07 |
Table 1: Physical test results of the three isolates.
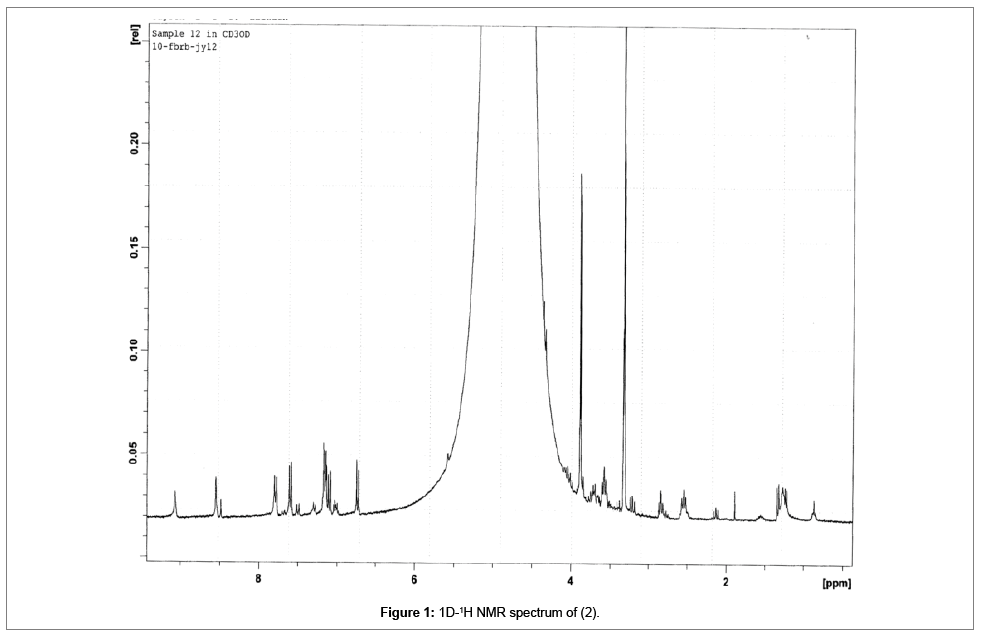
Figure 1: 1D-1H NMR spectrum of (2).
Structures were determined by physical (M.pt and Rf values), chemical (phytochemical screening), spectroscopic (1D-1H NMR, 2D-HSQC NMR and 2D-HMBC NMR) methods and comparison with literature data (Table 2) [11].
| 1H | δ (ppm) | |
|---|---|---|
| (2) | Ficuseptine-Cl | |
| 1 - (C-H2) | 3.59 | 3.54 |
| 3 -(C-H2) | 4.99 | 5.45 |
| 4 - | - | - |
| 5- (C-H) | 9.13 | 10.09 |
| 7- (C-H) | 8.60 | 8.20 |
| 4’OMe - (C-H3) | 3.88 | 3.80 |
| 4”OMe - (C-H3) | 3.89 | 3.80 |
Table 2:1H δ comparison of (2) with ficuseptine-Cl (4).
Structural elucidation
Ficuseptine (2) was obtained as second fraction from preparative chromatography of (F2). Vacuum drying gave orange-like paste (4.20 mg), M.pt 213-215 °C and Rf 0.14 with minor impurities [11].
Proton NMR assignments compared with literature values of ficuseptine chloride and showed exceptional resemblance, (Table 2).
The two methylene protons (2H-1) and (2H-3) resonate as triplets at δ 3.59 and δ 4.99 respectively. Electron dense δ 4.99 (2H-3) on (C- 3) depicts the strong electron withdrawing component, quaternary nitrogen (=N=+).
Protons resonating at δ 3.88 and δ 3.89 are singlets depicting two methoxy groups. Both δ 3.88 and δ 3.89 are para-aryls substituents. The methines δ 9.13 and δ 8.60 are singlets depicting two lone protons on the indolizidinium aryl component.
HMBC correlations of δ 33.1 from δ 2.55 (2J 2H-2) and δ 4.99 (3J 2H-3), δ 22.6 from δ 4.99 (2J 2H-3) and δ 60.9 from δ 2.55 (2J 2H-2) and δ 3.59 (2J 2H-1) is evident the methylene groups comprised in a discrete spin system. The H-C HMBC coupling pattern suggests that the methylene groups are interconnected in a planner system. The high chemical shift of methylene δ 60.9 (C-3) depicts connection to positively charged electronegative element (=N=)+.
Signal at δ 3.59 ppm shows HMBC correlation to the quaternary acetyl carbon, δ 155.3 (2J C-9). This signifies the three methylene groups, the quaternary nitrogen and carbon constitutes a five-membered ring system. Chemical shift at δ 155.3 ppm shows long range correlations from, δ 8.60 (3J 1H-7), δ 4.99 (3J 2H-3) and δ 9.13 (3J 1H-5). More so, δ 9.13 convinces that its bonding carbon is directly bonded to the quaternary nitrogen; hence, depicting that the five-membered spin system is joined adjacent to an aromatic ring (Figure 2).
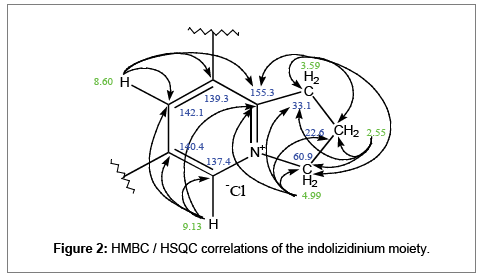
Figure 2: HMBC / HSQC correlations of the indolizidinium moiety.
2D NMR correlation analysis of 13C to 1H vice versa via HSQC and HMBC. nJ denoting long range correlations beyond 3J. Coupling constants J (Hz) were measured from 1D NMR spectra. Quaternary carbons were derived from correlation analysis of 1H to 13C via HMBC (Table 3).
| C | δ (ppm) | |||
|---|---|---|---|---|
| 13C | 1H –1J(HSQC) | 1H – 2J(gHMBC) | 1H –3J(gHMBC) | |
| 1 | 33.1 | 3.59, t, 1.8 Hz, 2H-1 | 2.55 (2H-2) | 4.99 (2H-3) |
| 2 | 22.6 | 2.55, quintet, 6.4 = 3.1Hz, 2H-2 | 4.99 (2H-3), 3.59 | - |
| (2H-1) | ||||
| 3 | 60.9 | 4.99, t, 1.6 Hz, 2H-3 | 2.55(2H-2) | 3.59 (2H-1) |
| 5 | 137.4 | 9.13, s, 1H-5 | 8.60 (1H-7) | |
| 6 | 140.4 | - | 9.13 (1H-5) , 8.60 (1H-7) | 7.80 (1H-2’) |
| 7 | 142.1 | 8.60, s, 1H-7 | 9.13(1H-5) | |
| 8 | 139.3 | - | 7.60 (1H-3”) | |
| 9 | 155.3 | - | 3.59 (2H-1) | 8.60 (1H-7), 9.13 (1H-5), 4.99 (1H-3) |
| 1’ | 126.8 | - | - | 8.60 (1H-7) |
| 2’ | 129.6 | 7.80, d, 1.8 Hz, H-2’ | - | - |
| 5’ | 115.8 | 7.14, d, 4.1 Hz,1 H-5’ | - | - |
| 1” | 127.7 | - | - | 8.60 (1H-7), 7.14 (1H-2”) |
| 2” | 130.9 | 7.15, dd, 8.2 = 4.1 Hz,1 H-2” | - | 7.60 (1H-3”) |
| 5” | 115.5 | 7.60, d, 1.7 Hz, 1H-5” | - | - |
| 4’OMe | 55.8 | 3.88, s-br, 3H-4’ | - | - |
| 4”OMe | 55.7 | 3.89, s-br, 3H-4” | - | - |
Table 3: 2D-HSQC and HMBC correlation assignments at selected proton and carbon systems of (2).
The two singlets resonating at δ 8.60 and δ 9.13 in the aromatic ring is likely in long range 1H-1H coupling at meta-meta positions. Their correlations to the quaternary δ 140.4 (2JC-6) and the presence of the other quaternary carbon δ 139.3 denotes ortho-para disubstituted carbons. Hence, depicting 1, 2, 3-trihydroindolizidinium moiety (Figure 3).
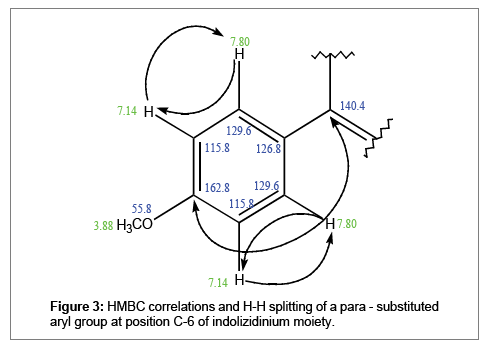
Figure 3: HMBC correlations and H-H splitting of a para - substituted aryl group at position C-6 of indolizidinium moiety.
1D-NMR showed two doublets at δ 7.80 and δ 7.60 ppm accounting for two protons each, whilst, the doublet of doublet at δ 7.14 accounts for four protons. 1H-1H coupling of δ 7.80 with δ 7.14 and δ 7.60 with δ 7.14 depict two para-substituted aryl groups.
δ 7.80 HMBC correlations to the quatenary δ 140.4 (3J1H-2’) and δ 162.8 (3J1H-2’) indicates that δ 129.6 is part of one para-substituted aryl bonded at position C-6 of the indolizidine moiety. δ 162.8 suggests the para-substiuent has high deshielding effect. Two protons resonating as dublets at δ 7.80 are likely on the ortho (2’), (6’) positions coupling to the doublet δ 7.14 at meta (3’), and (5’) positions. Singlet at δ 3.88 shows direct correlation to δ 55.8 and attributed as a methoxy group, hence, is the ideal para-substituent.
δ 8.60 HMBC correlations to the quaternary δ 127.7 (3JC-1’’) depicts association of the indolizidine moiety at position C-8 to the other parasubstituted aryl group. δ 7.14 HMBC correlations to δ 127.7 (3JC-2’’) and δ 162.1 (3JOMe-4’’) indicates that δ 129.6 is part of one para-substituted aryl bonded at position C-6 of the indolizidine moiety. δ 162.1 suggests the para-substiuent is a high shielding element. Two protons resonating as doublets at δ 7.60 are likely on the meta (3’’)-meta (5’’) positions coupling to the doublet of doublet δ 7.14 at ortho (2’’)-ortho (6’’) positions. Singlet at δ 3.89 shows direct correlation to δ 55.7 and attributed as a methoxy group, hence, is the ideal para-substituent (Figure 4).
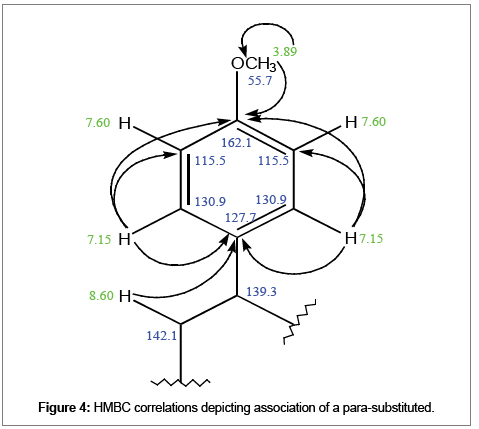
Figure 4: HMBC correlations depicting association of a para-substituted.
Main peak in the spectrum is m/z 332.1645 which matches exactly with the calculated ion for ficuseptine, C22H22O2N+, m/z 332.1645. The expected 13C signal is present at 333.1677, however, there is still a very small signal at 333.1714. Expected deuterated ion C22H21DO2N+, m/z 333.1708 is within 1 ppm of the peak observed at 333.1706 (Figure 5).
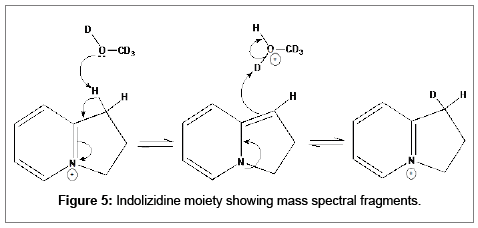
Figure 5: Indolizidine moiety showing mass spectral fragments.
The compound was established as ficuseptine chloride (2) on the basis of 1D 1H-NMR, 2D HSQC and HMBC spectroscopy, MS, melting point (213-215°C) and TLC analysis. Moreover, comparison with literature confirms the compound to be ficuseptine chloride (Figure 1), ruling out possibilities of N-oxides counter ions [11,12] (Figure 6).
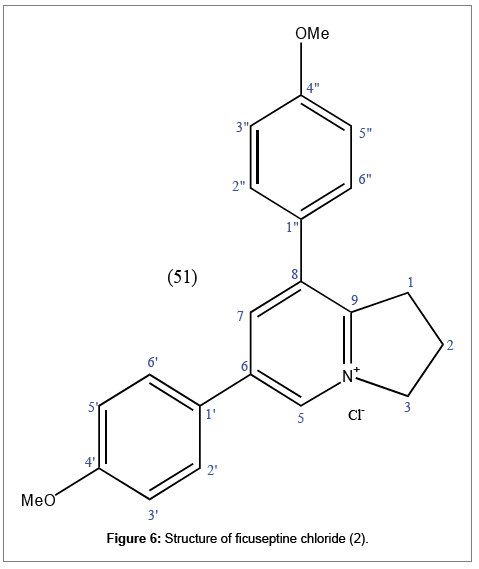
Figure 6: Structure of ficuseptine chloride (2).
Small quantity of fraction (1) was transferred to a NMR tube and dissolved in 1 ml CD3OD and analysed by Varian Mercury 300 MHz NMR at 10 scans. 1H nmr δ (CD3OD):9.07(s, 1H-5), 8.54 (s, 1H-7), 7.85 (d, J=1.8 Hz, H-2’ and H-6’), 7.60 (d, J=1.7 Hz, 1H-3” and 1H-5”), 7.15 (dd, 8.4=4.1 Hz, 1H-3’, 1H-5’, 1H-2” and ,1H-6”), 4.99 (t, J=1.6 Hz, 1H- 3), 3.89 (s-br, 3H-4”), 3.88 (s-br, 3H-4’), 3.57 (t, J=1.8 Hz, 1H-1) and 2.55 (quart, J=6.4, 3.1 Hz, 1H-2) ppm.
The compound was identified as ficuseptine (1) on the basis of its 1H-NMR spectral comparison with ficuseptinechloride (4) and other literature data (Rf and melting point) [11,12] (Figure 7).
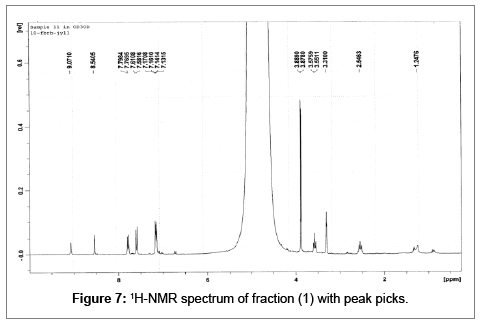
Figure 7: 1H-NMR spectrum of fraction (1) with peak picks.
Small quantity of fraction (3) was transferred to a NMR tube and dissolved in 1 ml CD3OD and analysed by Varian Mercury 300 MHz NMR at 10 scans. 1H nmr δ (CD3OD): 9.06(s, 1H-5), 8.52 (s, 1H-7), 7.77 (d, J=1.8 Hz, H-2’ and H-6’), 7.59 (d, J=1.7 Hz, 1H-3” and 1H-5”), 7.14 (dd, 8.4=4.1 Hz, 1H-3’, 1H-5’, 1H-2” and ,1H-6”), 4.99 (t, J=1.6 Hz, 1H-3), 3.89 (s-br, 3H-4”), 3.87 (s-br, 3H-4’), 3.56 (t, J=1.8 Hz, 1H- 1) and 2.54 (quart, J=6.4, 3.1 Hz, 1H-2) ppm.
The compound was identified as ficuseptine (1) on the basis of its 1H-NMR spectral comparison with ficuseptinechloride (4) and other literature data (Rf and melting point) [11,12] (Figure 8).
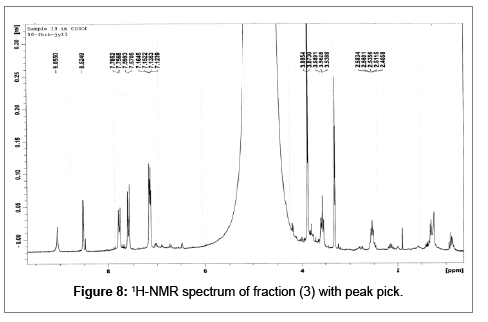
Figure 8: 1H-NMR spectrum of fraction (3) with peak pick.
The compound was identified as ficuseptine (3) on the basis of its 1H-NMR spectral comparison with ficuseptine chloride (2), ficuseptine (1) and other literature data (Rf and melting point) [11,12].
Spectroscopic assessment with 1H-NMR, 13C-NMR, IR and MS showed all fractions, (1), (2) and (3) to be ficuseptine. Fraction (2) was determined to be ficuseptine chloride based on 1H-NMR, 13C-NMR, IR and MS data. This is the first time this compound was reported in F. botryocarpa. With consistent spectral data fractions (1), (2) and (3) are ficuseptine bearing different anionic components. These ficuseptine derivatives are extremely polar and cationic. It was found that the fruit latex is not active in wet weather [6]. This may indicate that the stable ficuseptine is of bioactive significance vice versa.
More-over there appears an association between the gummy precipitate (F1) and the milky latex (FL) synonymous of the ficuseptine behaving as an emulsifying agent.
The ficuseptine derivatives are reported as chlorides, though the natural anionic form of the three isolated ficuseptine derivatives were not established. Further work is necessary to isolate the three ficuseptine derivatives in their natural form and confirm the emulsification claim.
The authors thank the PNG University of Technology Research Committee for funding the study. Dr. S. Akoitai, Applied Science, Department for bioassay and phytochemical screening, Mr. K. Aubeta, Bulolo University College for the plant identification and Prof. Juergen Reichardt, School of Pharmacy and Molecular Science, James Cook University, Australia for spectral analysis.
