Research Article - (2016) Volume 4, Issue 2
Aromatase, a catalyst in the aromatization reaction of androgens to estrogen, is a member of the cytochrome p450 superfamily, known as monooxygenases. The synthesized estrogen by Aromatase in breast cancer nourishes the cancer cells and assesses the hormonally growing of cancer cells. Therefore, Aromatase is considered as a potential target in treatment of breast cancer. Nowadays, major considerations in drug's selection in the treatment of cancers are shifted to natural sources due to their low toxicity profiles and better therapeutic functionality. In the present study, we have identified the binding modes of various xanthones, dietary supplements obtained from different plant sources, by using of efficient Biocomputational tools. Through docking studies, it is clear that 10 out of 13 ligands showing hydrogen bonds with amino acids like THR 310, PRO 249, ARG 113, GLY 439 and CYS 347. Sameathxanthone A showed highest dock score of -7.96 with the binding energy -38.46 kcal/mol and two hydrogen bonds with LEU 477 and VAL 373. Subsequently, the ADMET properties of compounds have been predicted computationally.
Keywords: Aromatase inhibitors; Natural products; Breast cancer; Xanthones; Docking score; ADMET properties
From age’s breast cancer, a type of ductal carcinoma is one of the most common cancers affecting female originating from the inner lining of the milk ducts of breast tissue [1]. Breast cancer alone comprises about 22.9% of all cancers in women. About 4.6 hundred thousand deaths were recorded worldwide in 2008 only due to breast cancer and it is about 13.7% of cancer deaths in women [2]. Many pathophysiological reasons like mutations in genes, inherited genetic predisposition, environmental, dietary and hormonal exposure leads to the development of breast cancer [3]. Many potential targets like Epidermal Growth Factor Receptor (EGFR), Vascular Endothelial Growth Factor Receptor (VEGFR), and enzymes like Aromatase, which catalyzes aromatization of androgens to estrogen, and also hormone receptors like estrogen receptor were identified for treatment of breast cancer [4]. The role of Aromatase in estrogen synthesis is depicted in Figure 1.

Figure 1: The role of Aromatase in oestrogen synthesis. The aromataseenzyme complex consists of a specific cytochrome P450 (CYP) heme protein in conjunction with a fluoroprotein, CYP reductase, localized primarily to the endoplasmic reticulum of ovarian granulosa cells in premenopausal women. Biologically significant aromatase activity in adipose tissue is the principal source of oestrogens after menopause. The synthesis of ovarian aromatase is regulated by an ovarian-pituitary (e.g., oestrogenfollicle- stimulating hormone (FSH)) feedback loop (www.mcdscape.com).
Aromatase Inhibitors (AIs), as a class of drugs in the treatment of postmenopausal women’s breast and ovarian cancer, are generally categorized into two groups: irreversible steroidal inhibitors and nonsteroidal inhibitors. The former, by binding to the active site, deals with permanently deactivating it, but the latter, by reversible competition for Aromatase enzyme with androgen, hinders binding androgens to the Aromatase enzyme [5]. The reversible Androgen-Aromatase binding inhibition contributes to the inhibition of androgens to estrogens conversions, in turn, preventing the steroidogenesis. It considerably reduces the circulation and the intra-tissue concentrations of estrogens. The reversible is more efficient than the permanent inhibition of Aromatase in chemotherapy of breast cancer, due to maintain the estrogen levels in normal rates approximately 70, 20 and 400 pmol/L for estrone, estradiol, and estrone sulfate, respectively in plasma for naturally postmenopausal women [6]. Figure 2 shows the mode of action of Aromatase inhibitor in Estron synthesis pathway.

Figure 2: The mode of action of Aromatse inhibitor in Estron synthesis pathway. Primary sites of selective (S) and nonselective (NS) blockade are indicated by heavy cross-mark. S=new aromatase inhibitors; NS=aminoglutethimide (www.mcdscape.com).
AIs can also be used to regulate Aromatase enzyme through other pathways and receptors such as, the modulation of Liver Receptor Homolog-1 (LRH-1) and orphan receptor. AIs regulate Aromatase in adipose tissue, testis, and granulose cells as well as contribute to over-expression of Aromatase in breast cancer patients [7]. Selective Aromatase modulators (SAMs) may be found based on the evidence for tissue-specific promoters of Aromatase expression [8].
With the clinical success of several synthetic Aromatase inhibitors (AIs) for the treatment of postmenopausal breast cancer, researchers have been investigating the potential of natural products as AIs [9]. The medicinal use of Natural Products (NPs) has a long history in treatment of various diseases. NPs have been utilized in the forms of herbal remedies, purified compounds, and precursor for combinatorial chemistry. The NPs can be considered for the inhibition of Aromatase due to the minimum side effects which can be as a result of natural product matrix components [9]. These components can be considered as the chemotherapeutic agents for the future clinical trials in breast cancer chemoprevention, treating postmenopausal breast cancer or preventing secondary recurrence of breast cancer [10]. It is also possible to synthetically modify of natural product scaffolds to enhance the inhibitory potential and improve the ADMET properties of such compounds [11].
Selection and preparation of protein
Aromatase structures with PDB ID-3EQM of obtained from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/) with X-ray diffraction resolutions of 2.90 Å. Preparation of the retrieved protein was performed by using protein Preparation Wizard of Schrodinger suite 2009 (Schrödinger Suite; Epik version 2.0; Impact version 5.5; Prime version 2.1, Schrödinger, LLC, New York, NY, 2009). The 3D structure of Aromatse is depicted in Figure 3.

Figure 3: The 3D structure of Aromatase.
Preparation of ligands
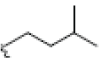
Preparation of ligands was done by using LigPrep 2.3 module of Schrodinger Suite 2009 using the OPLS force-field 2005 at biologically relevant PH. It performed by assigning the protonation states, including the disconnection of group I metals in simple salts, the deprotonation of strong acids and the protonation of strong bases, while adding explicit hydrogens and topological duplicates. The 2D structures of all compounds were presented in Table 1.
| N | Name | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | LPE | XPG | DG | HB-R | HB-D | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1-isomangostin | 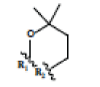 |
OH | H | H | OH | 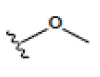 |
 |
2.527E+1 | -1.37 | 6.38 | -- | -- | ||
| 2 | 8-deoxygartanin | OH | 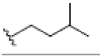 |
OH |  |
OH | H | H | H | 2.934E+1 | -2.46 | -9.70 | Thr310 | 2.051 | |
| 3 | 8-hydroxycudraxanthone G | OH |  |
 |
OH | H | H | OH | 3.616E+1 | -3.45 | -7.15 | Pro429 Thr310 | 1.914 1.912 | ||
| 4 | α-mangostin | OH |  |
OH | H | H | OH |  |
 |
4.542E+1 | -1.04 | 4.06 | Arg115 Pro429 Thr310 | 2.040 2.278 2.047 | |
| 5 | Cudraxanthone G | OH |  |
 |
 |
OH | H | H | H | 3.448E+1 | -2.17 | -13.02 | Thr310 | 2.127 | |
| 6 | Garcinone D | OH |  |
OH | H | H | OH | 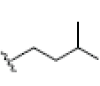 |
 |
3.335E+1 | -3.34 | -5.88 | Cys437 | 2.251 | |
| 7 | Garcinone E | OH | 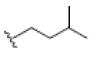 |
OH | H | 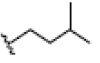 |
OH | OH | 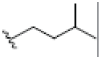 |
3.286E+1 | -0.40 | -13.91 | Arg115 Pro429 Thr310 | 2.099 2.207 2.091 | |
| 8 | Gartanin | OH | 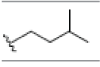 |
OH |  |
OH | H | H | OH | 3.400E+1 | -6.03 | 11.87 | Arg115 Thr310 | 2.388 1.843 | |
| 9 | Mangostinone | OH | 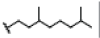 |
OH | H | OH | H | H | H | 2.960E+1 | -5.65 | -26.89 | Cys437 | 2.396 | |
| 10 | Sameathxanthone A | OH |  |
OH | H | OH | H | H | OH | 3.229E+1 | -7.96 | -38.46 | Met374 Leu477 | 2.145 1.804 | |
| 11 | Tovophylline | OH | 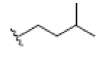 |
OH | H | 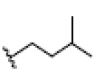 |
OH | 3.526E+1 | -4.49 | 4.97 | -- | -- | |||
| 12 | γ-mangostin | OH |  |
OH | H | H | OH | OH |  |
3.216E+1 | -5.12 | -16.70 | Gly439 Thr310 | 2.430 1.845 | |
| CONTROL | |||||||||||||||
| 13 | Dihydroisocoumarin 1 |  |
5.810E-1 | -5.28 | -14.29 | -- | -- | ||||||||
Table 1: Docking Scores and MMGBSA, Hydrogen bond residue numbers and distances of different Xanthone derivatives and Dihydroisocoumarin 1.
ADMET studies
The absorption, distribution, metabolism and excretion studies were performed via Qikprop 3.2 module of Schrodinger Suite 2009, the pharmacokinetics profiles of the compounds assessed by #start parameter, which indicates the number of property descriptors out of range for 95% of known drugs [12]. these criteria includes: SASA, FOSA, FISA, volume, PISA, Glob, Metab, QPlogKhsa, mol_MW, donorHB, accptHB, QPlogPo/w, QPlogPw, QPlogPoct‡, and QPlogPC16 [13,14], CNS [15], the human oral absorption level, the maximum transdermal transport rate (Jm), QPlogHERG, QPlogBB [16], QPPCaco [17], QPPMDCK, QPlogKp, IP(eV), EA(eV), and the number of violations of Lipinski’s rule of five [18] of the various Xanthone derivatives.
The toxicity of compounds was estimated via online TOPKAT approaches of Accelrys Environmental Chemistry and Toxicology Workbench, Accelrys Inc., San Diego, USA. TOPKAT has predicted the toxicity profiling of compounds, including Mutagenicity (Ames test v3.1), Rodent Carcinogenicity from the FDA dataset for both female and male (v3.1), Skin Sensitization (GPMT) (v.6.1), Skin Irritancy (v6.1), Ocular Irritation (v5.1), Weight Of Evidence (WOE) (v5.1), Developmental toxicity potential (DTP) (v5.1), Aerobic Biodegradability (v6.1), EC50, LD50, LC50, TD50 [19]. The ADME results are listed in Table 2.
| Molecules | #stars | #rotor | CNS | mol_MW | dipole | SASA | FOSA | FISA | PISA | WPSA |
|---|---|---|---|---|---|---|---|---|---|---|
| 1-isomangostin | 0 | 5 | 0 | 410.47 | 3.03 | 647.20 | 407.79 | 106.01 | 133.40 | 0 |
| Dihydroisocoumarin 1 | 0 | 6 | 0 | 306.36 | 4.59 | 595.38 | 411.23 | 87.85 | 96.29 | 0 |
| 8-deoxygartanin | 1 | 7 | -2 | 380.44 | 4.55 | 702.35 | 383.30 | 140.54 | 178.50 | 0 |
| 8-hydroxycudraxanthone G | 2 | 8 | -2 | 410.47 | 4.66 | 707.52 | 427.56 | 140.74 | 139.22 | 0 |
| a-mangostin | 2 | 8 | -2 | 410.47 | 4.67 | 745.61 | 470.53 | 131.58 | 143.50 | 0 |
| cudraxanthone G | 2 | 7 | -1 | 394.47 | 4.01 | 707.94 | 433.78 | 103.83 | 170.33 | 0 |
| garcinone D | 1 | 10 | -2 | 428.48 | 8.35 | 763.71 | 460.42 | 166.22 | 137.07 | 0 |
| garcinone E | 2 | 10 | -2 | 464.56 | 4.88 | 842.37 | 565.20 | 165.92 | 111.25 | 0 |
| gartanin | 1 | 8 | -2 | 396.44 | 5.17 | 706.99 | 381.57 | 176.56 | 148.86 | 0 |
| mangostinone | 1 | 8 | -2 | 380.44 | 3.99 | 717.68 | 327.96 | 167.46 | 222.26 | 0 |
| sameathxanthone A | 2 | 9 | -2 | 396.44 | 5.07 | 725.34 | 327.31 | 204.67 | 193.36 | 0 |
| tovophylline A | 2 | 7 | -2 | 462.54 | 4.99 | 818.31 | 537.02 | 143.68 | 137.61 | 0 |
| y-mangostin | 1 | 8 | -2 | 396.44 | 4.39 | 705.14 | 376.46 | 179.05 | 149.63 | 0 |
| Molecules | volume | Donor HB | Accpt HB | glob | QPpolrz | QPlog PC16 | QPlog Poct | QPlog Pw | QPlog Po/w | QPlogS |
| 1-isomangostin | 1228.78 | 2 | 5.50 | 0.86 | 41.09 | 11.78 | 19.37 | 10.15 | 3.99 | -5.19 |
| Dihydroisocoumarin 1 | 1047.34 | 0 | 5.75 | 0.84 | 32.83 | 9.23 | 13.56 | 6.75 | 2.83 | -3.51 |
| 8-deoxygartanin.mol | 1252.25 | 2 | 3.75 | 0.80 | 41.17 | 12.59 | 18.53 | 8.66 | 4.61 | -6.38 |
| 8-hydroxycudraxanthone G | 1298.22 | 1 | 3.50 | 0.81 | 41.99 | 12.49 | 17.21 | 6.47 | 5.17 | -6.58 |
| a-mangostin | 1330.98 | 2 | 4.50 | 0.78 | 43.35 | 13.08 | 19.51 | 8.98 | 4.82 | -6.74 |
| cudraxanthone G | 1288.11 | 1 | 3.75 | 0.81 | 42.53 | 12.37 | 17.35 | 6.90 | 5.29 | -6.66 |
| garcinone D | 1364.10 | 3 | 5.25 | 0.78 | 43.33 | 13.84 | 21.92 | 10.98 | 4.28 | -6.30 |
| garcinone E | 1530.07 | 3 | 4.50 | 0.76 | 49.72 | 15.33 | 23.19 | 10.25 | 5.58 | -7.92 |
| gartanin | 1267.48 | 2 | 3.50 | 0.80 | 40.86 | 12.78 | 18.45 | 8.25 | 4.52 | -6.38 |
| mangostinone | 1258.14 | 2 | 3.75 | 0.79 | 41.18 | 13.19 | 18.59 | 8.96 | 4.51 | -6.49 |
| sameathxanthone A | 1276.97 | 2 | 3.50 | 0.78 | 41.02 | 13.43 | 18.61 | 8.56 | 4.43 | -6.54 |
| tovophylline A | 1491.16 | 2 | 4.50 | 0.77 | 50.34 | 14.70 | 22.04 | 9.42 | 5.74 | -8.20 |
| y-mangostin | 1260.41 | 3 | 4.50 | 0.80 | 40.58 | 12.96 | 20.28 | 10.71 | 3.85 | -5.82 |
| Molecules | CIQPlog S | QPlog HERG | QPP Caco | QPlog BB | QPPMDCK | QPlog Kp | IP(eV) | EA(eV) | #metab | QPlog Khsa |
| 1-isomangostin | -6.18 | -4.48 | 978.71 | -0.70 | 483.34 | -2.52 | 8.97 | 0.47 | 7 | 0.65 |
| Dihydroisocoumarin 1 | -3.38 | -4.62 | 1454.76 | -0.63 | 741.84 | -2.22 | 9.56 | 0.62 | 3 | -0.13 |
| 8-deoxygartanin | -6.18 | -5.62 | 460.39 | -1.32 | 213.91 | -2.81 | 8.80 | 0.60 | 9 | 0.91 |
| 8-hydroxycudraxanthone G | -7.01 | -5.22 | 458.42 | -1.34 | 212.92 | -2.86 | 8.74 | 1.04 | 10 | 1.14 |
| a-mangostin | -6.51 | -5.70 | 559.95 | -1.34 | 264.32 | -2.67 | 9.01 | 0.54 | 10 | 0.94 |
| cudraxanthone G | -6.58 | -5.44 | 1026.34 | -0.92 | 508.81 | -2.16 | 8.79 | 0.68 | 9 | 1.10 |
| garcinone D | -6.48 | -5.76 | 262.83 | -1.87 | 116.70 | -3.14 | 9.08 | 0.64 | 9 | 0.71 |
| garcinone E | -7.48 | -5.91 | 264.56 | -1.94 | 117.53 | -3.23 | 8.68 | 0.52 | 13 | 1.28 |
| gartanin | -6.60 | -5.46 | 209.71 | -1.75 | 91.43 | -3.48 | 8.64 | 0.69 | 10 | 0.96 |
| mangostinone | -6.18 | -6.07 | 255.80 | -1.73 | 113.33 | -3.06 | 8.85 | 0.63 | 9 | 0.88 |
| sameathxanthone A | -6.60 | -5.94 | 113.51 | -2.19 | 47.09 | -3.75 | 8.78 | 1.00 | 10 | 0.93 |
| tovophylline A | -7.75 | -5.89 | 429.94 | -1.47 | 198.66 | -3.01 | 8.61 | 0.70 | 9 | 1.41 |
| y-mangostin | -6.11 | -5.48 | 198.62 | -1.78 | 86.22 | -3.52 | 8.72 | 0.55 | 10 | 0.66 |
| Molecules | OA | %OA | ro5 | ro3 | Jm | |||||
| 1-isomangostin | 3 | 100 | 0 | 1 | 0.008 | |||||
| Dihydroisocoumarin 1 | 3 | 100 | 0 | 0 | 0.563 | |||||
| 8-deoxygartanin | 1 | 100 | 0 | 2 | 0.000 | |||||
| 8-hydroxycudraxanthone G | 1 | 91.88 | 1 | 2 | 0.000 | |||||
| a-mangostin | 1 | 100 | 0 | 2 | 0.000 | |||||
| cudraxanthone G | 1 | 100 | 1 | 2 | 0.001 | |||||
| garcinone D | 1 | 95.29 | 0 | 2 | 0.000 | |||||
| garcinone E | 1 | 90.01 | 1 | 2 | 0.000 | |||||
| gartanin | 1 | 94.98 | 0 | 2 | 0.000 | |||||
| mangostinone | 1 | 96.43 | 0 | 2 | 0.000 | |||||
| sameathxanthone A | 1 | 89.69 | 0 | 2 | 0.000 | |||||
| tovophylline A | 1 | 94.75 | 1 | 2 | 0.000 | |||||
| y-mangostin | 1 | 90.64 | 0 | 2 | 0.000 |
Recommended range: CNS: -2-2, SASA: 300-1000, FOSA: 0-750, FISA: 7-330, Volume: 500-2000, PISA: 7-200, Glob: 0.75-0.95, IP(eV): 7.9-10.5, EA(eV): -0.9-1.7, Metab: 1-8, QPlogS: -6.5-0.5, CIQPlogS: -6.5-0.5, QPlogHERG: >-5, QPPCaco: 25-500, QPlogBB: -3.0-1.2, QPPMDCK: 25-500, QPlogKp: -8- -1, QPlogKhsa: –1.5-1.5, HOA: 1-3, mol_MW: 130-725, donorHB: 0.0-6.0, accptHB: 2.0-20.0, QPlogPC16: 4.0-18.0, QPlogPoct‡: 8.0-35.0, QPlogPw: 4.0-45.0, QPlogPo/w: -2.0-6.5, OA (Human Oral Absorption): 1-3, %OA (Percent Human Oral Absorption): 0-100%
Table 2: ADME Profiling.
Receptor-Ligand interactions
Docking studies were performed by using Glide 5.5 module in Extra Precision (XP) mode [20-22] and the molecular mechanics/ Generalized Born Surface Area (MMGBSA) [23] for interaction of each ligand-protein complex has been calculated via Prime 2.1 application of Schrodinger Suite 2009. The results of docking and MMGBS are available in Table 1. Then, the complex of receptor-ligand was mapped via XP visualizer approaches of Schrodinger 2009 and the receptor surfaces were configured based on the electrostatic potential of residues in the binding packet of protein by truncating the receptor surface in 5Å from ligand with 20% transparency. The pose of ligand is visualized via Ligand Interaction Diagram module of Schrodinger 2009.
Preparation of the receptor and ligands
Aromatase PDB structure has been downloaded from RCSB Protein Data Bank (http://www.rcsb.org/pdb/). Then via ProPrep wizard of Shrodinger 2009, the protein preparation was carried out by removing the disulphide and trisulphide bonds of a protein, all water molecules except those which bind through hydrogen bonds with the residues in the binding side of a protein and adding hydrogen’s to the molecule to satisfy the valences of the molecule. It employed OPLS 2005 force-field with RMSD as 0.30. The binding site of proteins has been detected by the pose of ligand presented in receptor crystal which was extracted from PDB structure.
In this study, 12 different Xanthone derivatives have been investigated and compared with a coumarin based selective Aromatase inhibitor derivatives, Dihydroisocoumarin 1 with IUPAC ID: [(3R,4R)-(-)-6-methoxy-1-oxo-3-pentyl-3,4-dihydro-1H-isochromen- 4-yl acetate] as the control. All ligands were prepared by using Ligprep wizard of Schrodinger 2009 under biologically related PH using Epik approach and OPLS2005 force-sfield (optimized potentials for liquid simulations force field as a model for environmental force-field in the body) [24]. The stereoisomer has generated at most 32 combinations per ligand. The potential energies of ligands are presented in Table 1.
Docking calculations using Schrodinger 2011
All ligands were docked in the active site of Aromatase (PDB ID:3EQM) with binding pocket including Met374, Val 373, Phe134, Arg115, Ile133, Ile132, Cys437, Ala438, Phe148, Gly439, Leu152, Met303, Ile442, Phe203, Met160, Met446, Ser199, Ala307, Ala306, Thr310, Trp224, Val370, Ser478, Le477 and Leu372 amino acids via GLIDE ver. 5.5 of Schrödinger software suite 2009. Docking was performed using extra precision (XP) docking and scoring, flexible docking option to generate conformations internally during the docking process and add Epik state penalties to docking score options. The binding energies of all ligand-receptor complexes have been evaluated using prime_mmgbsa ver. 1.3 and the complexes were visualized by XP visualizer module of Glide ver. 5.5, based on Glide XP_GScore.
According to the affinity values, binding energy and number of hydrogen bonds with the active site of receptor presented in Table 1, Sameathxanthone A possessed the highest docking score -7.96, with the binding energy -38.46 and 2 hydrogen bonds to Leu477 and Val 373 with bond distance 1.804 Å and 2.145 Å respectively. The values of the Docking scores, binding energies, number of hydrogen bonds and bond distances of all 12 Xanthone derivatives and control compound were presented in Table 1.
Figure 4 shows the complex of Sameathxanthone A with the binding site of Aromatase, in which H-bonds indicated with yellow dotted line and the values of bond distances with pink color. The binding packet was created by using Create Binding Site Surfaces option by truncating receptor surface at 5.0 Å from the ligand, using the surface transparency 25% with solid style and electrostatic potential color scheme.

Figure 4: The complex of Sameathxanthone A with the binding site of Aromatase, yellow dotted line: H-bonds and the values of bond distances in Å.
Figure 5 shows the 2D structure of Sameathxanthone A pose in the binding pocket of Aromatase with residue numbers and chemical characterization of residues in which the cyan, green and purple colors indicate polar, hydrophobic and charge positive amino acids respectively. The hydrogen bond is shown by the pink line.

Figure 5: The 2D structure of Sameathxanthone A pose in binding pocket of Aromatase with residue numbers and chemical characterization of residues; Cyan: hydrophobic, Green: polar, Purple: charge positive amino acids, and Pink line: hydrogen bond.
ADMET investigation
For designing a druggable molecule, it is necessary to check the drug ability of ligands in terms of oral and intestinal absorption level, the ability of ligands to distribute through the blood stream, level of metabolism, the ability of excretion from the body besides their toxicity profiling. For estimation of drug pharmacokinetics and metabolism, QikProp software investigates around 24 molecular descriptors, which determine the #star parameter. The recommended values for #star by Schrodinger is a range 0-5 for the computed property of a molecule out of the range for 95% of known drugs, including MW, dipole, IP, EA, SASA, FOSA, FISA, PISA, WPSA, PSA, volume, #rotor, donorHB, accptHB, glob, QPpolrz, QPlogPC16, QPlogPoct, QPlogPw, QPlogPo/w, logos, QPLogKhsa, QPlogBB, #metabol.
The likely oral availabilities of the compounds have been evaluated using [16] MW ≤ 500 Da, accptHB ≤ 10, donorHBD ≤ 5, QPlog Po/w ≤ 5, #rotor ≤ 10 [25] which are known as the Lipinski’s criteria. The oral absorption or the likelihood of the oral availability is computed by using parameters including log S>-5.7, QPPCaco>22 nm/s and # primary metabolites<7 which are known as the Jorgensen’s famous ‘‘Rule of Three’’ (ro3). The Jotgensen’s ro3 beside human oral absorption percentage, the predicted qualitative human oral absorption, the predicted of human gut barrier absorption, QPPCaco, and CIlog S are used to predict the bioavailability [26]. The permeability prediction depends on the molecular properties such as the size, flexibility which depends on the #rotor [27], overall lipophilicity, shape, and the capacity to make hydrogen bonds.
The QPlogBB is used to predict the blood brain barrier permeability for each compound and accessibility of bio actives for central nervous system. It has contrary relation to the polarity of the compound. For CNS availability and activity of compounds, in addition of OPlogBB, the CNS activity, and QPPMDCK also have to be considered. The recommended range for CNS activity, QPlogBB and QPPMDCK are (-2-+2), (-3-1.2) and (25-500 nm/s) respectively.
The QplogKp is known as the skin permeability parameter. It is used to predict the penetration of drugs/compounds through the skin. For the maximum transdermal transport rates, Jm, predicted by the equation (1):
Jm=Kp × MW × S (1)
Where Kp is the skin permeability obtained from QPlogKp, MW is molecular weight and S is the aqueous solubility obtained from QPlogS and Jm is the prediction of the maximum transdermal transport rate in μg cm-2hr-1.
Another parameter which is used for prediction of availability of drugs for their target is Prediction of binding to human serum albumin. QPlogKhsa is used to predict the ability of compounds to bind to the plasma proteins such as globulins, human serum albumin, lipoprotein and glycoprotein. The plasma proteins binding ability is directly influencing the drug efficacy, the distribution of drug through the blood stream and the availability of drugs for their target. The plasma-proteins binding tendency of a drug has an inverse relation to the target availability. Therefore, the less degree of plasma-protein binding is desirable for designing drug. The recommended range for QPlogKhsa is -1.5-1.5 for 95% of known drugs.
The #metab parameter is known as the number of likely metabolic reactions which is necessary for determining the level of accessibility of compounds to their target sites after entering into the blood stream. The recommended range of #metab is 1-8.
QPlogHERG is an important parameter to predict cardiac toxicity of compounds. It is used to predict the IC50value for blockage of Human Ether-a-go-go-Related Gene Potassium (HERG K+) channel. HERG K+ plays role in the electrical activity of the heart during systolic and diastolic periods by encoding the potassium ion (K+) channel. This channel has also modulating function in the nervous system [28]. The Blockage of HERG K+ channel is potentially toxic for the cardiac and nervous system [29]. The recommended range of IC50 values for blockage of HERG K+ channels is QPlogKhsa >-5.
The ADME investigation of Sameathxanthone A showed that all parameters except #metab score10, CIQPlogS score 6.6 and QPlogeHERG score 5.9 were in the recommended range of druglikeness, pharmacokinetics and metabolism criteria. Among all, only one of Xanthone derivatives, 1-isomangostin, showed as same ADME profiling as Dihydroisocoumarin 1 and possessed all ADME parameters within the recommended range. However, 1-isomangostin showed very low the affinity value, around -1.37, for inhibition of Aromatse. Figure 6 depicted the comparison of ADME criteria between Dihydroisocoumarin 1 and Sameathxanthone A.

Figure 6: The comparison of ADME criteria between Dihydroisocoumarin 1 and Sameathxanthone A.
Toxicity
Some important parameters for toxicity investigation have been predicted via online TOPKAT approaches of Accelrys Environmental Chemistry and Toxicology Workbench. The carcinogenicity of compounds have been predicted based on structural similarity between compounds and structures available in both the FDA (U.S. Food and Drug Administration) and NTP (National Toxicology Program) databases for male and female of rat and mouse (FR, FM, MR and MM). According to the predicted carcinogenicity based on the FDA database, Sameathxanthone A is non-carcinogenic for both female and male mouse and male rat, but weak carcinogen for a female rat with probability 0.34, and based on NTP database, it is safe for both male and female rat and female mouse but carcinogen for male mouse with probability 0.66. It is not skin and ocular irritant, but strong skin sensitizer with probability 0.83. According to WOE (weight of evidence for rodent carcinogenicity), it showed no carcinogenic activity in rodents. Based on the DTP (developmental toxicity potential), it was toxic, and got a positive discriminant score with the probability 0.62 for the toxicity, but showed no genotoxicity or mutagenicity potential. The Rat Oral LD50 value evaluated as 1.32 gm/kg, which was within the Optimum Prediction Space (OPS) and indicated the higher safety of this compound. The half of effective concentration values for Daphina Magna model indicated that the EC50 value of Sameathxanthone A, 0.22 mg/l, was within the recommended range. The half of the lethal concentration value (LC50) was around 0.0001 g/l and the half of the tolerance doses for carcinogenicity by feeding for mouse and rat were 31.003 and 93.278 mg/kg_body_weight/day respectively. It is liable for degradability through aerobic-biodegradability.
In this paper, Schrodinger 2009 and online Topkat approaches of Accelry were employed for in silico investigation of the pharmacodynamics and pharmacokinetics of the 12 xanthone based molecules with target “Aromatase” in breast cancer. The affinity value of 12 Xanthone derivatives has been compared with each other and with the affinity value of Dihydroisocoumarin 1 (IUPAC ID: [(3R, 4R)-(-)-6-methoxy-1-oxo-3-pentyl-3,4-dihydro-1H-isochromen-4-yl acetate]) as a selective Aromatase inhibitor. The result indicated that Sameathxanthone A with -7.96 kcal/mol and the binding energy -38.46 possessed significantly higher docking score than the rest of Xanthones and Dihydroisocoumarin 1and it can be introduced as a candidate for breast cancer treatment. It showed a perfect toxicity profile, but still further study is necessary to improve the ADME properties of Sameathxanthone A.
There is no conflict of interest among the authors.
The authors are grateful to Mr. Raghuraj the C/O Aravinda Biosolution Ltd., for providing financial support and encouraging throughout.
