Research Article - (2018) Volume 6, Issue 1
Received Date: Nov 20, 2017 / Accepted Date: Dec 20, 2017 / Published Date: Jan 02, 2018
A new cerebroside (2R)-2-hydroxy-N-[(Z,2S,3S,4R)-1-O-β-D-glucopyranosyl-3,4-dihydroxynonadec-8-en-2-yl] nonacosanamide (1) was isolated from the wood of roots of V. paradoxa along with six known compounds including catechin (2), quercetin (3), spinasterol 3-O-β-D -glucopyranoside (6), gallic acid (7) and a mixture of β-sitosterol (4) and stigmasterol (5). The structure of the new compound was established by 1D (1H and 13C NMR) and 2D NMR (COSY and HSQC) spectroscopy, extensive mass spectrometry and by comparison with published data. The antibacterial, α-glucosidase and alkaline phosphatase (AP) inhibitory activities of all the pure compounds were evaluated. The antibacterial activities were evaluated against three gram negative bacteria (Escherichia coli; Salmonella typhimurium and Pseudomonas aeruginosa) while APs inhibitory activities were evaluated on h-TNAP and h-IAP. Significant antibacterial activity was recorded for quercetin (3) against P. aeruginosa. Most of the compounds except 1 and 6 were found to be inhibitors of α-glucosidase. The highest inhibitory potential being recorded for quercetin (3) with IC50 value of 4.30 ± 0.01 μM, 55 fold higher than the standard drug acarbose (IC50=234.6 ± 2.01 μM). All tested compounds exhibited moderate inhibitory activities against APs. h-TNAP inhibitory values were ranged between 41.24 ± 1.33 μM and 312.54 ± 6.44 μM while h-IAP inhibitory values were in the range of 47.95 ± 0.35 μM and 777.47 ± 18.55 μM. Quercetin (3) was found to be the most active h-IAP inhibitor (IC50=47.95 ± 0.35 mM), whereas, spinasterol 3-O-β-D-glucopyranoside (6) was found to be the most active h-TNAP inhibitor (IC50=41.24 ± 1.33 mM). The new compound (1) showed moderate inhibition on h-IAP (78.11 ± 3.70 μM) and on h-TNAP (88.84 ± 2.70 μM).
Keywords: Vitellaria paradoxa; Sapotacea; Cerebroside; Antibacterial activity; α-glucosidase; Alkaline phosphatase
Sphingolipids are a class of lipids containing a backbone of sphingoid bases, a set of aliphatic amino alcohols and found essentially in all animals, fungi, as well as some prokaryotic organisms and viruses. The most commonly occurring sphingoid bases from plant sources are phytosphingosine [1]. Most of the sphingoid bases in natural products are N-acylated with long-chain fatty acids to produce ceramides(s). The fatty acids of ceramide vary in chain length (14 to 30 carbon atoms), degree of unsaturation (but are mostly saturated), and presence or absence of a hydroxyl group on the α- or ω-carbon atom [2]. This variation in chain length, the presence or the absence of double bonds and hydroxyl groups, including their position, the stereochemistry, the glycosidation and the phosphorylation produce a large structural diversity in sphingolipids. More than 300 different types of complex sphingolipids have been reported [2]. Cerebrosides or monohexosylceramides, are simple members of the glycosphingolipids. Their structures are characterized by the presence of a sphingoid base backbone that is amide-linked to a fatty acid, and carbohydrate head group (mostly glucose or galactose) attached to C-1 hydroxyl group of the sphingoid base. Sphingolipids, particularly cerebrosides exhibit various biological activities such as antiulcerogenic activity [3], antifungal, antitumor, immunomodulating, antiviral, antitumor, immunostimulatory activities [1,4], anti-neuroinflammatory activity [5], antiplasmodial, antileismanial and cytotoxic activities [6]. Therefore, sphyngolipids could be considered as an extremely interesting group of natural products that should continue to attract attention for further investigations that may lead to new drug candidates.
Infectious, chronic and degenerative diseases are nowadays the major health problems in developing as well as developed countries as the impacts of these diseases are immense and can be felt across the world. They are major causes of morbidity and death [7]. Bacterial infections and typhoid fever are among the major diseases with high degree of risk within the population [8], due to the increasing prevalence of multi-resistant strains of bacteria and the recent appearance of strains with reduced susceptibility to antibiotics that raises the spectrum of untreatable bacterial infections and adds urgency to the search for new infection-fighting strategies.
Diabetes mellitus (DM) is a chronic metabolic disorder resulting from impaired glucose homeostasis. It has been stated that the number of patients with the hyperglycemic condition is expected to increase to 642 million by 2040 [9]. The inhibition of α-glucosidase (EC. 3.2.1.20) is among the most important therapeutic approaches in treating diabetes [10]. α -Glucosidase inhibitors slow down the process of digestion and absorption of carbohydrates resulting to the reduction of the peak concentration of postprandial blood glucose and the blood sugar level. Most of the available α-Glucosidase inhibitors have been clinically associated with serious side effects [9,10]. Therefore, there is a great necessity to search for alternatives α-glucosidase inhibitors without side reactions.
Alkaline phosphatase (AP; EC 3.1.3.1) is a kind of enzyme found in human body, precisely in liver, digestive system, kidneys, bile ducts, placenta and bones. It is an enzymes that facilitates the absorption of the nutrients across the cell membrane catalyzing their dephosphorylation and serve as a source of inorganic phosphate. There are four APs isozymes in human body among which three are tissue specific: placental AP (PLAP), germ cells AP (GCAP), and intestinal AP (IAP). The fourth one is tissue-nonspecific AP (TNAP), present in bone, liver, and kidney it is 50% alike to the three others [11]. Among all these isozymes, TNAP and IAP are highly structurally similar. TNAP is mainly involved in the hydrolysis of pyrophosphate to inorganic phosphate thus maintaining the bone mineralization process and growth, while IAP plays an important role in the lipid absorption across the small intestine and detoxification of Lipopolysaccharides, an endotoxin produce by gram negative bacteria, responsible for inflammation [11]. Elevated or lowered level of this enzyme will result in the development of various pathophysiological conditions, for example an over expression of APs can cause calcification, osteoarthritis, carcinogenis, diabetes, etc. In 2009, a study revealed that increased levels of APs and phosphate are independent risk factors for all-cause and cardiovascular mortality in the general population [12]. Individuals with a combination of increased APs and phosphate levels had the highest mortality risk. Modulation of APs is a potential novel treatment strategy that might reduce vascular calcification and improve cardiovascular outcomes in patients with diabetes mellitus type 2 [13]. The development of potent inhibitors is therefore required for their treatments. The synergistic effects of phytomedicine make traditional medicine promising therapeutic options against infectious and chronic diseases [14-16].
Vitellaria paradoxa is the only species of the genus Vitellaria which belongs to the Sapotaceae family represented by 58 genus with about 120 species. Commonly known as shea butter tree, V. paradoxa is largely distributed in the semi-arid zone of sub-Saharan Africa from Senegal in the west to Uganda [17]. As far as its applications are concerned, the plant is used as food and medicine. Thus in the Adamawa region of Cameroun where it is known as Karehi in Fufulde , its root barks are used against breast cancer [18]. It is also mentioned all over the world in the treatment of several infectious diseases such as skin infections, diarrhoea, digestive disorders, dysentery, convulsions, cough, leprosy, malaria, jaundice [19,20]. Its medicinal properties are often attributed to its secondary metabolites, since the plant is known as a source of biologically active compounds including terpénoïdes such as α-amyrin, α-amyrin acetate, β-amyrin, β-amyrin acetate, β- sitosterol, stigmasterol, lupeol and lupenone, phenolic compounds as gallic acid, catechin, epicatechin, epicatechin gallate, gallocatechin, epigallocatechin, gallocatechin gallate, and epigallocatechin gallate e s as well as quercetin and trans-cinnamic acid [21,22]. Recently we reported the isolation and structural elucidation of a new ursan-type triterpenoid from the stem bark of V. paradoxa [23]. In a continuation of our investigation of chemical constituents from this plant, we describe here the isolation and structural elucidation of a new phytosphingosine-type cerebroside, (2R)-2-hydroxy-N-[(Z ,2S ,3S , 4R )-1-O-β-D-glucopyranosyl-3,4-dihydroxynonadec-8-en-2-yl] nonacosanamide (1, Figure 1), from the wood of roots of this important medicinal plant. To the best of our knowledge this is the first report of a study on the wood of roots of V. paradoxa .
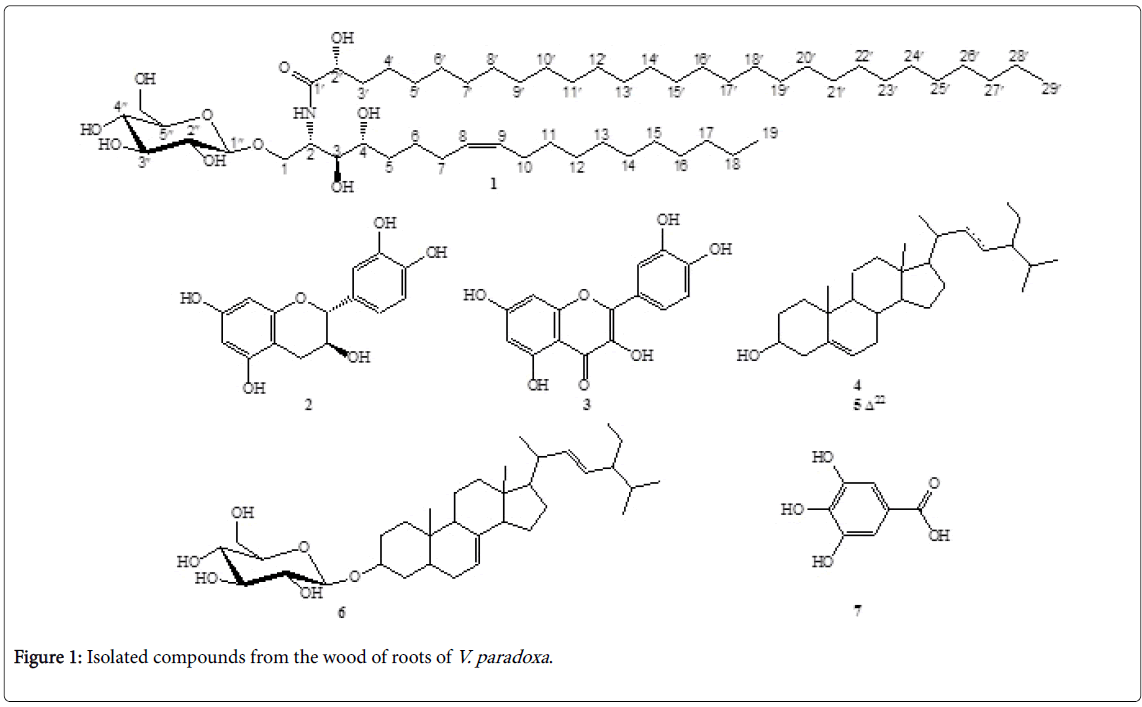
Figure 1: Isolated compounds from the wood of roots of V. paradoxa .
General experimental procedure
Column chromatography (CC) was performed on silica gel 60 (70-230 mesh, Merck), monitored by analytical Thin layer chromatography (TLC) on silica gel precoated plates F-254 Merck (20 × 20 cm). Spots were visualized under UV light (254 and 365 nm), sprayed with 20% sulfuric acid then heated at 105°C for 15 min. The melting point of the new compound was recorded in an open capillary using Stuart melting point apparatus (SMP-3) and is uncorrected. ESIMS spectra (ionization voltage 3 kV) were registered on a Q-TOF Ultima spectrometer (Waters). A spectrometer Bruker Avance AV-500 (125 and 500 MHz) was used for NMR spectra. CDCl3 or DMSO-d6 was used as solvents and TMS as internal standard. Ciprofloxacin (Sigma-Aldrich, St Quentin Fallavier, France), oxytetracycline (Pantex, Hollande) and Ketoconazole (European Pharmacopoeia (EP) Reference Standard, Sigma-Aldrich), were used as reference antibiotics whereas Thiazolyl Blue Tetrazolium Blue (MTT) from Sigma Aldrich was used as bacterial growth indicator for antimicrobial assay. α- Glucosidase from Saccharomyces cerevisiae recombinant G0660-750UN, 4-nitrophenyl α-D-glucopyranoside ≥ 99% N1377-1G, Acarbose (positive control) and Sodium dihydrogen phosphate were purchased from Sigma Aldrich, (Germany). 100% dimethylsulfoxide (DMSO) was purchased from LAB-SCAN (Thailand). Solvents used were of analytical grade and water was distilled.
Plant material
The woods of roots of V. paradoxa collected in April 2016 at Ngaoundere, Adamawa Region (Cameroon), was authenticated at the National Herbarium of Yaounde (Cameroon) where a voucher specimen (50216/HNC) is deposited.
Extraction and isolation
Dried vegetal material (1 kg) was extracted by maceration at room temperature using MeOH (10 L) for 72 h and the solution afterwards filtered and concentrated under vacuum resulting after drying to a brown powder extract (189.6 g) which was then partitioned with EtOAc till exhaustion. The EtOAc soluble portion was subjected to column chromatography (CC) on silica gel (hexane/EtOAc/MeOH gradient of increasing polarity) to afford compound 1 (6 mg) at AcOEt/MeOH 98:2, catechin (2, 120 mg) at AcOEt 100%, and quercetin (3, 75 mg) at AcOEt/MeOH 95:5 together with four fractions V1-V4 on the basis of TLC composition. Fraction V1 (0.36 g) was subjected to CC on silica gel and eluted with a gradient of increasing polarity using Hexane/EtOAc (1:0-0:1, v/v) to yield a mixture of phytosterols (23 mg): β-sitosterol (4) and stigmasterol (5) at hexane/ EtOAc 90:10. Fraction V2 (0.58 g) was submitted to CC of silica gel with EtOAc as solvent of elution in isocratic mode yielding spinasterol 3-O-β-D -glucopyranoside (6, 7 mg). Fraction V3 (2 g) was passed through a silica gel CC eluted with a gradient of hexane/EtOAc (7:3-0:1) to give gallic acid (7, 18 mg) at hexane/EtOAc 2:8 by crystallization from MeOH.
(2R)-2-hydroxy-N-[(Z,2S,3S,4R)-1-O-β-D-glucopyranosyl-3,4- dihydroxynonadec-8-en-2-yl] nonacosanamide (1): White powder, m.p. 147-149°C. -TOF-ESI-MS+: m/z =927.4 [M]●+ calculated for C52H97NO12. Important 1H-1H COSY correlations are illustrated in Figure 2. 1H NMR (500 MHz, DMSO-d6): δH: 0.84 (t , J=6.5Hz, 6H, 3H-29', 3H-18), 1.20-1.34 (br s, H-11-18, H-4'-28'), 1.49 (m , H-3'a), 1.50 (m , 2H, H-6), 1.60 (m , H-3'b), 1.92 (m , 2H, H-5), 1.94 (m , 2H, H-7), 1.98 (m , 2H, H-10), 2.95 (td , J=8.9, 2.7 Hz, 1H, 2"-H), 3.04 (dd, J=9.1, 3.7 Hz, 1H, H-4"), 3.09 (dd, J=5.8, 2.0 Hz, 1H, H-5"), 3.15 (td, J=8.8, 3.0 Hz, 1H, H-3"), 3.31 (m , 1H, H-4), 3.38 (m , 1H, H-3), 3.44 (m , 1H, H-6"a), 3.66 (m , 1H, H-1a), 3.67 (m , 1H, H-6"b), 3.81 (dd , J=10.0, 6.7 Hz, 1H, H-1b), 3.86 (dd , J=10.0, 6.4 Hz, 1H, H-2'), 4.10 (dt , J=15.5, 5.3 Hz, 1H, H-2), 4.15 (d , J=7.7 Hz 1H, H-1"), 4.35 (m , 1H, OH-5"), 4.52 (t , J=5.6 Hz, 1H, OH-6"), 4.77 (t , J=5.6 Hz, 1H, OH-3), 4.91 (t , J=4.3 Hz 2H, OH-3", OH-4"), 4.95 (d , J=3.5 Hz, 2H, OH-2"), 5.31 (m , 1H, H-9), 5.37 (m , 1H, H-8), 5.60 (d , J= 4.9 Hz, 1H, OH-4) 7.54 (d , J=9.5 Hz, 1H, NH). -13C NMR (125 MHz, DMSO-d6): δC: 13.9 (C-29', C-19), 22.1 (C-18, C-28'), 24.4 (C-4'), 25.5 (C-6), 26.5 (C-10), 26.6 (C-7), 28.6-29.2 (C-12-16, C-5'-26'), 31.3 (C-17, C-27'), 32.0 (C-11), 32.3 (C-5), 34.3 (C-3'), 49.8 (C-2), 61.0 (C-6"), 68.9 (C-1), 69.9 (C-4"), 70.5 (C-4), 70.9 (C-2'), 73.4 (C-2"), 74.4 (C-3), 76.5 (C-3"), 76.9 (C-5"), 103.3 (C-1"), 129.8 (C-9), 130.2 (C-8), 173.7 (C-1').
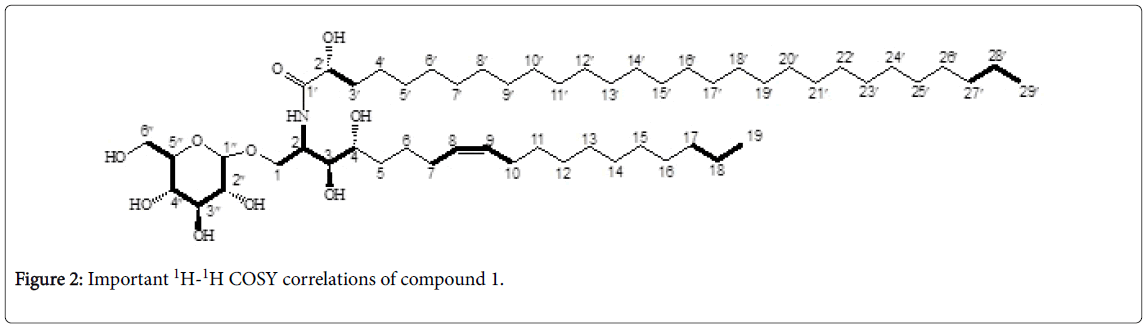
Figure 2: Important 1H-1H COSY correlations of compound 1.
in vitro antibacterial assay
Microorganisms and culture media: Compounds were tested against 4 microorganisms, four bacterial strains (Salmonella typhi ATCC6539, Salmonella typhimurium, Pseudomonas aeruginosa ATCC9721, Escherichia coli ). Among these microorganisms E. coli were isolated from Ayub Theacthing Hospital of Abbottabad (Pakistan) while S. typhimurium was isolated from the Laboratory of Bacteriology and Mycology of the “Centre Pasteur” of Yaounde, Cameroon respectively. These microorganisms were maintained on agar slant in refrigerator at 4°C. The reference strains were obtained from American Type Culture Collection (ATCC). Mueller Hinton Agar (MHA) for the activation of tested Gram-negative bacteria while Mueller Hinton Broth (MHB) was used for the in vitro antibacterial assay as culture media [24].
MTT colorimetric assay for MIC and MBC determination: The microdilution method using the MTT colorimetric assay was used to determine the Minimal Inhibitory Concentrations and Minimal Bactericidal Concentrations (MICs and MBCs) of the samples and reference antibiotics. Indeed, samples were first dissolved in 50 μL of DMSO 10%. To the solution obtained 950 μL of MHB was added and then serially diluted two fold in a 96-well microplate. The final concentration of DMSO was lower than 2.5% in order to not affect the microbial growth. 100 μL of inoculum (1.5 × 106 CFU.mL-1) in MHB were then added. The plates were covered with a sterile plate sealer, then agitated with a plate shaker to mix the contents of the wells and incubated at 37°C for 18 h. Wells containing MHB broth, 100 μL of inoculum and DMSO to a final concentration of 2.5% served as negative control. The assay was done in triplicate and the MIC of each sample was detected after 18 h of incubation at 37°C following addition of 40 μL MTT (0.2 mg.mL-1) and incubation at 37°C for 30 min. Viable bacteria converted the yellowish solution of MTT to a dark blue characteristic of the water-insoluble MTT formazan by mitochondrial dehydrogenases of living cells. The MIC was the sample concentration that prevented this color change of the medium and exhibited complete inhibition of microbial growth; in others words, the MIC corresponds to the lowest concentration of the compounds that inhibits visible growth (visual turbidity) that was detected by the reduction of MTT into MTT formazan, a dark blue molecule. The highest dilution of a compound in which no a dark blue color appears corresponds to its MIC. For the determination of the MBC, 50 μL aliquots of the preparations (from the well which did not show any growth after incubation during MIC assays) were added to 150 μL of adequate broth. These preparations were incubated at 37°C for 48 h. The MBC was considered as the lowest concentration of samples that prevented the color change of the medium after addition of MTT as mentioned above [25,26].
α-Glucosidase inhibitory activity: The α-glucosidase inhibition assay was performed spectrophotometrically using p-nitrophenyl-α-Dglucopyranoside (p-NPG) as substrate in a 96-well microtiter plate (Corning Costar, Cambridge, MA, USA), following the reported procedures [27] slightly modified. Briefly, the assay started with the pre-incubation at 37°C of a mixed solution containing 70 μL phosphate buffer (70 mM, pH 6.8), 10 μL of test compound (0.1 mM) and 10 μL of enzyme (α-glucosidase (2.5 U.mL-1). after 5-10 min of preincubation, 10 μL of p-NPG (10 mM) was added to each well of a 96 well plate and further incubated at 37°C for 20 min. The reaction was stopped by adding 80 μL of 0.2 M Na2CO3 solution. The solutions of α- glucosidase and its substrate were prepared in 70 mM phosphate buffer (pH 6.8), while the tested compounds were dissolved in DMSO 10%. Negative control was constituted of 10 μL of distilled water instead of test compounds, while acarbose was used as positive control. The activity of test compounds against glucosidase isoenzymes was determined by measuring the amount of p-nitrophenol released at a wavelength of 405 nm using a FluoStar Omega plate reader (BMG LabTechnologies, Offenburg, Germany). The percent inhibition was calculated using the following equation:
% inhibition=[100-(Absorbancetest well/Absorbancecontrol)] × 100
For all the test compounds, assay was performed in triplicate. IC50 values of the compounds showing more than 50% activity were determined by further serial dilution into eight different concentrations. IC50 values were determined using GraphPad PRISM software version 5.0 (San Diego, CA, USA) as the mean ± S.E.M. of three assays.
Alkaline phosphatase inhibition assay: Alkaline phosphatase activity was performed using a previous reported luminescence method [28] with slight modifications in which CDP-Star was used as substrate for the determination of enzyme inhibition of compounds on bovine kidney alkaline phosphatase (h-TNAP) enzyme and calf intestine alkaline phosphatase (h -IAP). Initial screening was performed for all the tested compounds at a concentration of 0.1 mM. An assay buffer constituted of 8 M diethanolamine (DEA), 2.5 mM MgCl2 and 0.05 mM ZnCl2 at pH 9.8, was used. The assay mixture containing 10 μL of tested compound (0.1 mM) and 20 μL of enzyme h -TNAP (1:800 times diluted (0.8 units per mL) enzyme in assay buffer) or 20 μL of h -IAP (1:800 times diluted (1 unit per mL) enzyme in assay buffer) was preincubated for 4 to 5 minutes at 37°C, the luminescence was then measured as a pre-read using microplate reader (BioTek FL × 800, Instruments, Inc. USA). After that, 20 μL of CDP-star (final concentration of 110 μM) was added to initiate the reaction and the assay mixture was incubated again for 15-20 min more at 37°C and then, the change in the luminescence was measured as after-read. The inhibitory activity of each tested compound was compared with negative control (without any inhibitor), while Levamisole (2 mM per well) and L-phenylalanine (4 mM per well) were used as a positive controls respectively against h -TNAP and h -IAP. For all the compounds which exhibiting more than 50% inhibition of either h - TNAP or h -IAP activity, further analysis were performed in order to determine IC50 values. For this aim, serial dilutions (6-8 concentrations) of each compound (100 μM to 20 nM) were prepared in assay buffer and their dose response curves were obtained by assaying each inhibitor concentration on both Aps isozymes using the above mentioned reaction conditions. All experiments were performed in triplicate then the IC50 values were determined by the non-linear curve fitting program PRISM 5.0 (GraphPad, San Diego, California, USA).
Statistical analysis: All values obtained were compared using Oneway ANOVA with a Student Newman-Keuls post-test using GraphPad Prism 6 Software, Inc. (San Diego, CA, USA). Differences at p<0.05 were considered statistically significant.
Compound 1 was obtained as a white powder, its molecular formula C52H97NO12 was deduced from the TOF-MS ESI+ analysis which showed a molecular ion peak at m/z 927.4 [M]+ consistent with the molecular formula C52H97NO12. Subsequently, the structure was fully elucidated by 1H and 13C NMR spectroscopy. The 1D NMR spectrum pattern of compound 1 coincided with glycosphingolipids skeleton, which shows the presence of an amide linkage, a sugar, and long chain aliphatic moieties. Indeed, the two signals at δC 49.8 (C-N) and 173.8 (C-O) in the 13C NMR spectrum of compound 1 together with the signal at δH 7.54 (1H, d, J=9.5 Hz) can be attributed to the signals of an amide function -NHCO- suggesting the presence of an amide group. The signal at δH 4.15 (1H, d, J=7.7 Hz) that is associated to the carbon at δC 105.6, can be attributed to the anomeric proton of a sugar moiety. All these signals combined with the ones of two long chain aliphatic moieties including aliphatic methylenes (δH 1.20-1.36, m) and a triplet of six protons (δH 0.86, 6H, t, J=6.9 Hz), suggested a glycosphingolipid nature of compound 1 [29-33]. The presence of a sugar moiety was confirmed by a positive reaction of compound 1 in the Molish test. The 13C NMR spectrum also shown a set of C-atom signals appeared at δC 61.0 (CH2), 69.9 (CH), 73.4 (CH), 76.4 (CH), 76.8 (CH), and 103.4 (CH), that suggested the presence of a β-glucopyranoside. The high coupling constant of the anomeric proton (J=7.7 Hz) corresponding to two trans diaxial protons, further confirmed the β-configuration of the glucoside unit. The position of the glucose moiety was found to be at C-1 due to the downfield chemical shift observed for the hydroxymethylene carbon C-1 at δC 68.9 [34,35]. Further extensive analysis of 1H NMR spectra revealed two olefinic proton signals at δH 5.37 (m , H-8), and 5.31 (m , H-9) attributable to the presence of a disubstituted double bond. Furthermore, it is known that the geometry of the double bond in a long-chain alkene can be determined on the basis of the 13C-NMR chemical shifts of the methylene carbons adjacent to the olefinic carbons. Usually, the chemical shifts for the adjacent carbons to a cis (Z) double bond appear in the range of δC 26-28, while those of a trans double bond appear in the range of δC 32-33 [35,36]. Thus, the Δ8 double bond was determined to be cis (Z) due to the downfield chemical shifts of C-7 (δ 26.6) and C-10 (δ 26.5) [37].
1H NMR spectrum of 1 also showed three other methine signals at δH 3.38 (m, CHOH), 3.34 (m, CHOH), 3.86 (dd ; 10.6; 6.7, CHOH), attributed to H-3, H-4 and H-2' respectively and were supported by the signals at δC 70.5 (C-4, CHOH), 70.9 (C-2', CHOH), and 74.4 (C-3, CHOH) in the 13C NMR spectrum. The above data suggested that the sphingoid base of compound 1 is phytosphingosine [38]. Therefore, compound 1 is suggested to be a molecular species of phytosphingosine-type cerebroside possessing 2-hydroxy fatty acid and β-glucopyranose moieties. It has been stated that the chemical shift of the H-2 signal and the 13C chemical shifts of C-1-C-4, C-1' and C-2' of glucosphingolipids are especially suitable for the determination of the absolute stereochemistry of the phytosphingosine moiety [30,39]. Literature survey revealed that the absolute configuration at C-2 of most natural cerebrosides is all-2S; the biogenetic pathway of the natural cerebrosides suggested a 2S, 3S-configuration of the phytosphingosine part [40-44]. Thus, from the chemical shift of H-2 (4.10, dt ; J=15.5; 5.3 Hz) and the carbon chemical shifts at δC 68.9 (C-1), 49.8 (C-2), 74.4 (C-3), 70.5 (C-4), 173.8 (C-1') and 70.9 (C-2') and all the facts above mentioned, the configuration of compound 1 was essentially identical to those reported for other cerebrosides with (2S, 3S, 4R)-phytosphingosine and (2R)-2-hydroxy fatty acid moieties [45,46].
The 1H-1H COSY spectrum presented a correlation between the amide proton at δH 7.51 and 2-H methine at δH 4.10 which in turn coupled to three other protons at δH 3.81, 3.38, and 3.62 corresponding respectively to H-1a, H-3 and H-1b. Furthermore, H-3 (δH 3.38) showed correlation with H-4 (δH=3.31). Cross peaks were also observed between the signal at δH 3.86 (H-2') and the downfield proton signals at δH 1.50 corresponding to 2H-3'. This confirmed the presence of the fourth hydroxyl group at C-2' of the fatty acid chain. The positions of the three hydroxyl groups in the long chain base were further confirmed by the mass fragmentation pattern (Figure 3). The COSY spectrum also revealed the correlations between the protons at δH 0.86 with a set of protons at δH 1.26 that in turn correlated with others protons at δH 1.34, in addition with the correlations between all the protons of the sugar moiety. The coorelation between the two olifenic protons H-8 at δH 5.37 and H-9 at δH 5.31, as well as the correlations of these protons and the adjacent ones at δH 1.94 (C-7) and δH 1.98 (C-10) respectively were also visible.
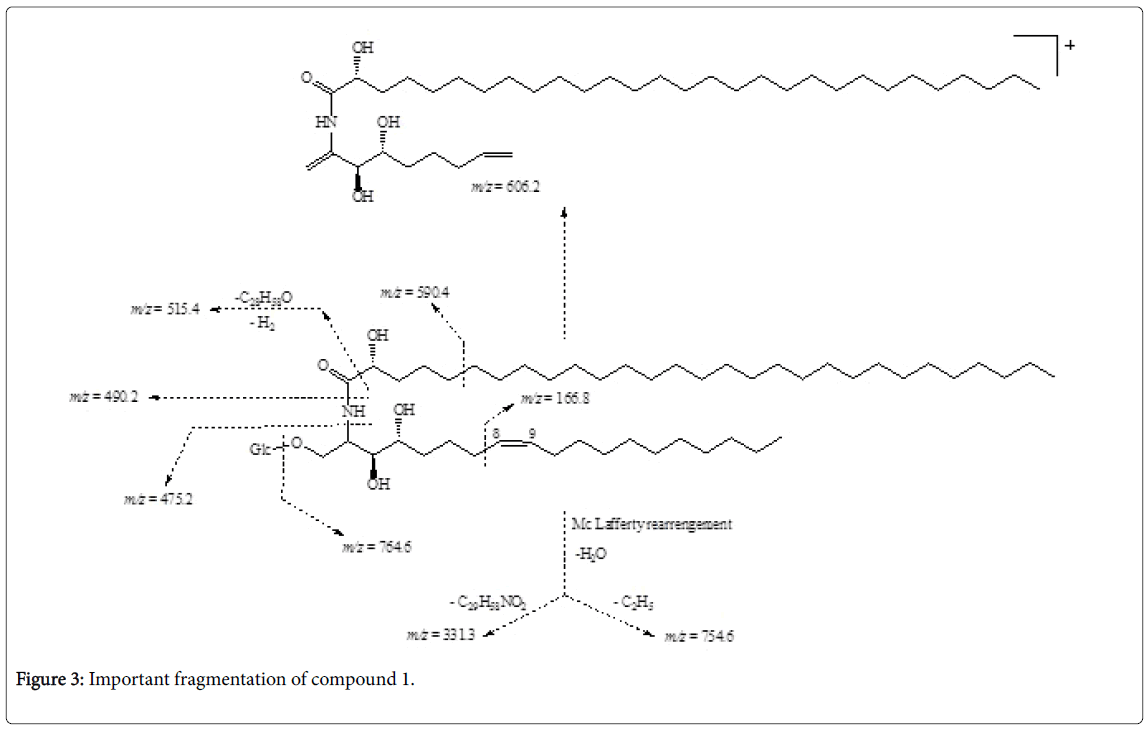
Figure 3: Important fragmentation of compound 1.
The length of the long chain base was determined by the characteristic ion at m/z 475.2 [Glc-OCH2CHCH(OH)CH(OH)(CH2)3CHCH(CH2)9CH3]+ (Figure 3). The fragment at m/z =166.8 [CH3(CH2)9CHCH]+ resulting from the cleavage in α of the Δ8 double bond confirmed the position of the double bond in the long chain base. The typical fragment ions at 764.6 [M-163]+ and at 606.2 [M-163]+ (Figure 3), resulting from the loss of the sugar moiety followed by the cleavage in α of the Δ8 double (between C-9 and C-10), further confirmed the presence of the glucoside moiety and the ester function. The length of the fatty acid chain was deduced from the fragment at 515.4 [GlcOCH2C(=CH2)NHCOCH(OH)(CH2)9CHCHCO]+ (Figure 3), resulting from the cleavage in α of the carbonyl group of the amide function. From the foregoing data, the structure of compound 1 was elucidated as (2R)-2-hydroxy-N-[(Z,2S,3S,4R)-1-O-β-Dglucopyranosyl- 3,4-dihydroxynonadec-8-en-2-yl] nonacosanamide and named Vitellaroside. To the best of our knowledge, this is the first time that a cerebroside is reported from Vitellaria paradoxa .
The known compounds (2-7) (Figure 1) were identified by comparing their NMR spectroscopic data with those reported in the literature, as catechin (2), quercetin (3), β-sitosterol (4), stigmasterol (5), spinasterol glucoside (6) and gallic acid [22].
Antibacterial activities
All pure compounds were screened for their antibacterial activities using the microbroth dilution method in 96-well micro-plates against three gram negative (-) bacteria: Escherichia coli, Salmonella typhimurium and Pseudomonas aeruginosa. The antibacterial activity was measured as MIC which is defined as the concentration that inhibits the growth of 50% of organisms. The results presented in Table 1, indicated that all the tested compounds were active depending on the bacterial strain with MIC values ranging from 8 to 128 μg/mL. Compound 7 inhibited the growth of 100% of the 3 tested bacteria with the MICs ranged from 16 to 128 μg/mL. The antibacterial activity of compounds has been defined as significant when MIC < 10 μg/mL, moderate when 10
| Bacterial strains | Tested compounds and MIC values (µg/mL) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 6 | 7 | Ciprofloxacin | ||||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | ||
| E. coli | Isolate | - | - | - | - | 16 | 128 | 128 | - | 64 | - | 0.5 | 0.5 |
| S. typhi | ATCC6539 | - | - | 128 | 128 | - | - | 64 | - | 64 | 128 | 0.5 | 1 |
| Isolate | 64 | - | 64 | - | - | - | - | - | 32 | 64 | 0.5 | 0.5 | |
| P. aeruginosa | ATCC9721 | 32 | 128 | 16 | 128 | 8 | 32 | 64 | 128 | 16 | 128 | 0.5 | 1 |
Table 1: MICs and MBCs of isolated compounds on the selected bacteria.
All the bacterial strains used in this study were gram-negative bacteria possessing complex and multilayered lipopolysaccharides cell walls. Therefore, for many compounds including synthetic and natural antibiotics, the access to this membrane is more restricted [48]. The activity of all the compounds against the gram (-) bacteria used in this study suggests that these compounds could be able to cross this tough barrier. All these compounds presented in their structures many organic functions (hydroxyls groups, amide, alkene, ester functions, phenols and carboxylic acids) that can be responsible for the observed activities [49,50]. This study confirms the fact that the number and the position of hydroxyl groups influenced the membrane interaction effects of organic compounds. Antibacterial activity of the isolated compound obtained in this study may provide some explanation for the traditional uses of V. paradoxa .
α-Glucosidase and alkaline phosphatase inhibitory activities
The pure isolated compounds were tested for their glucosidase inhibitory potential by in vitro enzyme assay. Following preliminary screening, catechin (2), quercetin (3) and gallic acid (7) showed very strong inhibitory effects against α-glucosidase, and were further studied for their concentration dependent activity in order to calculate IC50 values (Table 2). Spinasterol 3-O-β-D -glucopyranoside (6) and the new compound 1 showed a weak activity, 35.62% and 41.42% respectively. Quercetin (3) and gallic acid (7) possessed high potency with the IC50 value of 4.30 ± 0.01 and 5.35 ± 0.18 μM, respectively, as compared with that of acarbose (IC50=234.6 ± 2.01 μM) which was used as a positive control. In addition, compound 2 also exhibited a potent activity against α-glucosidase with an IC50 of 68.3 ± 1.25 μM which was ~ 4-fold higher compared to the positive control (acarbose). Catechin (2), quercetin (3) and gallic acid (7) may be the main antihyperglycemic agents present in this plant, and they have been already reported for their glucosidase inhibition activities [51,52].
| Code | α-Glucosidase | Alkaline | |
|---|---|---|---|
| h-TNAP | h-IAP | ||
| IC50 (µM) ± SEM or % Inhibition | |||
| 1 | 41.42 % | 88.84 ± 2.70a | 78.11 ± 3.70a |
| 2 | 68.3 ± 1.25a | 312.54 ± 6.44b | 777.47± 18.55b |
| 3 | 4.30 ± 0.01b | 121.44 ± 3.51c | 47.95 ± 0.35c |
| 6 | 35.62 % | 41.24 ± 1.33d | 68.91 ± 2.19a |
| 7 | 5.35 ± 0.18b | 236.39 ± 4.82e | 81.89± 4.73a |
| Acarbose | 234.6 ± 2.01c | / | / |
| Levamisole | / | 20.2 ± 1.9f | / |
| L-Phenylalanine | / | / | 80.2 ± 0.001a |
Table 2: α-Glucosidase and alkaline phosphatase inhibition of the isolated compounds.
The luminescence of the solutions of active Aps gives a possibility to evaluate the inhibitory activity of some compounds when exposed to these solutions by attenuation of the luminescence. This activity can therefore be measured via a luminescence based assay. In this study, the isolated pure compounds from the wood of roots of V. paradoxa were investigated for their potential to inhibit the human tissue nonspecific (h-TNAP) and intestinal (h-IAP) alkaline phosphatase isozymes.
The results resumed in Table 2 shows that all tested compounds were active against both isoenzymes of alkaline phosphatase. h-TNAP inhibitory values were ranged between 41.24 ± 1.33 and 312.54 ± 6.44 μM while h-IAP inhibitory values were in the range of 47.95 ± 0.35 and 777.47± 18.55 μM. Most of the tested compounds (60%) display selective inhibitory activity for h-IAP. Among all the tested compounds, quercetin (3) showed the highest activity against h -IAP which was found to be 47.95 ± 0.35 nM, ~ 2 fold more active than the standard L-phenylalanine and ~ 3 fold more selective for h-IAP over h-TNAP. Meanwhile, spinasterol 3-O-β-D-glucopyranoside (6) was found to be the most potent inhibitor of h -TNAP through the series, but its activity was ~ 2 fold less when compared to the one of Levamisole used here as the standard for this isoenzyme. Catechin (2) was found to be the least inhibitor of both isoenzymes h -IAP and h-TNAP with IC50 values of 777.47 ± 18.55 μM and 312.54 ± 6.44 μM respectively. The new compound 1 also demonstrated a good inhibition potential with a slight specific inhibition against h-IAP.
In the present study, we reported the isolation a new cerebroside, Vitellaroside (1), from the wood of barks of Vitellaria paradoxa, together with catechin, quercetin, spinasterol 3-O-β-Dglucopyranoside, gallic acid and a mixture of phytosterols. All the pure compounds were tested for antibacterial, α-glucosidase and alkaline phosphatase inhibitory activities and, most of them show either moderate or low activities. Therefore, some like quercetin and gallic acid were very active against α-glucosidase. To the best of our knowledge, (2R)-2-hydroxy-N-[(Z,2S,3S,4R)-1-O-β-Dglucopyranosyl- 3,4-dihydroxynonadec-8-en-2-yl] nonacosanamide (1) is the first cerebroside identified from this plant and from the Sapotaceae family. Except the new compound (1), all the others have already been reported from this plant.
Authors are thankful to the Centre of Advanced Drug Research (CADR), COMSATS Institute of Information Technolgy (CIIT), Pakistan, for research facilities and the Third World Academy of Sciences (TWAS), Italy for financial support under the TWAS-CIIT Post Graduate Sandwich program [grant No. 3240287163, 2015] granted to JNN. Authors are also thankful to the Medical Microbiology laboratory, Pasteur Centre, Yaoundé (Cameroon) and to the Ayub Theacthing Hospital of Abbottabad (Pakistan) for providing some clinical bacteria.
No potential conflict of interest was reported by the authors
